
Abstract
In order to develop preclinical models of malignant astrocytomas and oligodendrogliomas, a series of 54 resected gliomas (37 from oligodendroglial lineage and 17 from astrocytic lineage) were xenografted subcutaneously into nude mice. Molecular alterations commonly observed in gliomas subtypes, including LOH 1p and 1q, LOH 19q, LOH 10p and 10q, LOH 9p, TP53 and PTEN mutations, EGFR amplification, CDKN2A homozygous deletion and telomerase reactivation were systematically screened in the original and xenografted tumours. In all, 23 gliomas grew in nude mice. The most anaplastic tumours were selected as shown by pathological and molecular studies of the original tumour as well as shorter survival in patients whose tumours were successfully grafted. Comparison between the two growth profiles showed that 10q LOH and EGFR amplification gave a tumorigenic advantage. With a few exceptions, the genetic pattern was remarkably stable before and after growth in nude mice.These results suggest that subcutaneous xenografts are useful and reproducible models to analyse the molecular profile of malignant astrocytoma and oligodendroglioma. This represents the first step to improve our understanding of the correlations between molecular alterations and response to standard or experimental therapies.
Similar content being viewed by others


Main
Over the last decade, important advances have been made in the understanding of the molecular pathways involved in the progression of astrocytomas and oligodendrogliomas (Louis et al, 2001). Recent data also suggest that acquired genetic alterations, such as LOH 1p–19q, could be important predictors of prognosis and response to therapy (Cairncross et al, 1998; Smith et al, 2000; Ino et al, 2001). Unfortunately, the lack of clinically relevant models of gliomas seriously affects fine analysis of the correlation between genotype and treatment response. This is particularly clear for oligodendrogliomas, which are very difficult to maintain in in vitro cultures (Merrill and Matsushima, 1988; Post and Dawson, 1992; McLaurin et al, 1995). In this setting, well-characterised xenografts of gliomas could be of help, as specified by the recommendations of the European Medicines Evaluation Agency (EAEMP, 1999) and the EORTC Laboratory Research Division (EORTC LRD, 2001). However, it remains to be demonstrated that astrocytic and oligodendroglial tumours can grow reproducibly in nude mice while maintaining the same profile of genetic alterations as in the original tumour.
Material and methods
Tumour classification
A set of 54 gliomas was surgically resected at the Pitié-Salpêtrière hospital, Paris and the Centre Hospitalier Régional, Nancy. An informed consent was obtained for all patients. After pathological review according to the current WHO guidelines (MK) (Kleihues and Cavenee, 2000), except for the addition of a recently individualised subgroup of glioblastoma with an oligodendroglial component (GBMO) (He et al, 2001), the tumours were classified as oligodendroglioma (one O), mixed gliomas (two OA), anaplastic oligodendroglioma (29 AO), anaplastic oligoastrocytoma (one AOA), four GBMO and finally a subset of 17 glioblastoma (GBM) that was used as a control group.
Xenograft protocol
Each tumour was xenografted subcutaneously into the scapular area. Following a maximal delay of 2 h after surgical resection, small fragments (∼50 mm3) of tumour tissue were xenografted onto at least three anaesthetised nude mice (Swiss nu/nu) aged 6–8 weeks. Before xenograft, tumours were kept in foetal calf serum-free DMEM (Sigma-Aldrich, St Louis, MO, USA). The mice were maintained under clean room conditions and received sterile rodent food and water ad libitum. Their care and housing were in accordance with institutional guidelines as put forth by the Ministère de l'Agriculture et de la Forêt, Direction de la Santé et de la Protection Animale, Paris, France and the standards required by the UKCCCR guidelines (Workman et al, 1998).
The mice were kept alive for a maximum of 12 months until the growth of the xenografts. The tumours were measured once a week in two perpendicular dimensions and their volumes were estimated using the formula (width)2 × (length)/2. Latency was defined as the time between first transplantation and the appearance of a palpable tumour. The doubling time was calculated once it became stable, that is, usually after the fifth passage. The mice were killed when the tumour volume reached 2000 mm3 and the xenografts were maintained by serial transplantation.
DNA extraction
Paired blood and tumours could be obtained for 31 initial tumours, blood could not be obtained for 12 tumours, and unfortunately tumour samples were not fresh frozen in 11 cases. Blood DNA was extracted using the Nucleon BACC3 DNA Extraction kit (Amersham Bioscience, Piscataway, NJ, USA) and tumoral DNA was extracted using the QIAamp DNA minikit, as described by the manufacturer (Qiagen, Venlo, NL, USA).
Microsatellite analysis for loss of heterozygosity (LOH) on chromosomes 1, 9p, 10 and 19q
Blood and tumour DNA were screened for LOH on chromosome 1p, using the following polymorphic markers: D1S450, D1S2667, D1S234, D1S255, D1S2797, D1S2890, on chromosome 1q using D1S2878, D1S249, D1S2785 markers. On chromosome 9p LOH was screened, using the following markers: D9S286, D9S168, D9S1870, D9S156, D9S1687, spanning the region located near CDKN2A. On chromosome 10p, LOH was screened using the following markers: D10S249, D10S189, D10S547 (near hTR repressor), D10S585 (near hTR repressor), D10S548, D10S204, on chromosome 10q using D10S537, D10S219, D10S1744 (near PTEN), D10S541, D10S579, D10S1755, D10S1671, D10S597, D10S1693, D10S209, D10S587 (near DMBT1), D10S1723, D10S212, D10S537, D10S541, D10S597, D10S1693, D10S212 markers spanning the region located between 10q21.22 and 10qter. And for chromosome 19q, LOH was screened using the following markers: D19S425, D19S219, D19S888, D19S412, D19S418. One of the primers was labelled with the Hex, Fam or Ned fluorochromes (Applied Biosystems, Foster City, CA, USA). The samples were run on an automatic sequencer and analysed with the Gene Scan program (Applied Biosystems, Foster City, CA, USA).
Screening of the PTEN/MMAC1 and TP53 gene mutations
PTEN/MMAC1 mutations were screened by the denaturing gradient gel electrophoresis (DGGE) technique in the entire coding sequence of the nine exons and their corresponding splice junctions using previously described primers (Zhou et al, 1999). TP53 mutations were screened by the DGGE technique for exons 5–8 and their using previously described primers (Hamelin et al, 1994). DNA showing altered DGGE profiles were sequenced bidirectionally using the Perkin Kit and sequencer. When a DNA variant was found, the corresponding blood DNA was sequenced in order to differentiate somatic events from constitutional variants (polymorphism or germline mutation).
Screening of EGFR gene amplification and P16/CDKN2A gene homozygous deletion
EGFR amplification was screened by semiquantitative PCR using primers and protocol described previously (Hunter et al, 1995). P16/CDKN2A homozygous deletions were screened by semiquantitative PCR using primers and protocol previously described (Walker et al, 1995).
Screening of telomerase activity
Telomerase activity was screened by Telomeric Repeat Amplification Protocol (TRAP) technique using the TRAPeze kit (Intergen Co, Purchase, NY, USA) as described by the manufacturer.
Statistical. analyses
Descriptive statistics for continuous variates are provided as mean, standard deviation, median, minimum and maximum. For categorical variates, frequency distribution, median, minimum and maximum are provided.
The relation between categorical variates is described using the χ2 method, or Fisher's exact test when the χ2 method is not appropriate.
Prognostic factors for tumour growth delay are identified using univariate analysis (log rank test) and multivariate analysis fitting Cox's proportional hazard regression models. All tests are considered significant at the 0.05 significance level. Odds ratios are presented with their 95% confidence interval.
Analyses were performed using the SAS/STAT software v8.0 (SAS Institute, Inc., Cary, NC, USA).
Results
Patient characteristics and relationship with tumour growth
The 54 surgical samples were obtained from 33 men and 21 women, with a median age of 57. 4 years (range 20–77). The post-op median survival was 11.03 months after a median follow-up of 31.9 months (range 1–37.2).
Out of 54 tumours, 23 were established in nude mice (42.2%). In total, 14 were derived from oligodendroglial tumours (one AOA, nine AO, four GBMO) and nine from GBM. When a tumour was tumorigenic, it grew on at least 60% of mice at the first transplantation and on 80–100% mice on later transplantations.
None of the three low-grade gliomas were established in nude mice, while 23 out of 51 high-grade gliomas did (45%).
Out of 21 grade IV gliomas, 13 were established in nude mice (62%), while 10 out of 30 grade III did (33%) (P=0.0047).
Patients, whose tumours did not grow had a longer survival (14.9 months) (range 1.4–37.2) than those whose tumour could be established (9.1 months) (range 0.2–32.5) (P=0.0003).
Histology after xenograft
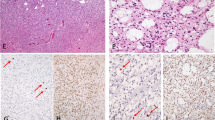
After xenograft in nude mice, tumours presented a dedifferentiated phenotype devoid of any particular pattern with small round or fusiform cells. Grafted oligodendrogliomas displayed a complete loss of ‘honeycomb’ appearance (Figure 1B, D). The cellular density was very high. Neither endothelial hyperplasia nor endothelial proliferation could be observed. Many mitotic and apoptotic figures were seen in the same samples, while central areas of necrosis were almost constant. Tumours lost their glial differentiation whatever their initial type (oligodendroglial or astrocytic), except one tumour with an initial morphology of mixed anaplastic glioma, which exhibited an oligo-like differentiation after xenograft (ABE).

Representative histology of two tumours before and after xenograft on nude mice. (A) GIR in situ tumour, anaplastic oligodendroglioma morphology, haematoxylin–eosin HE staining (magnification × 200); (B) ODA-17-GIR corresponding xenograft, passage 1, dedifferentiated morphology, HE staining (× 400); (C) ROM in situ tumour, GBM histology, haematoxylin–eosin (HE) staining (× 200); (D) GBM-7-ROM corresponding xenograft, passage 3, dedifferentiated morphology, HE staining (× 400).
Genetic alterations of primary tumours and relationship with tumour growth
As summarised in Table 1, screening for p53 mutations, PTEN mutations, EGFR amplifications and p16/INK4a homozygous deletions was performed in 43 tumours. Screening for LOH on chromosomes 1p, 1q, 19q, 10p, 10q, 9p was performed for 31 tumours and for telomerase activity in 22 tumours.
Tumours were separated into two groups. The first one consisted of tumours that grew in nude mice for at least two transplantations (n=19) and the second group of those that did not (n=24). Comparison of the two groups showed that growth in nude mice was correlated to 10q LOH and EGFR amplification (P=0.01 and 0.0035, respectively). In addition, when genetic alterations linked to anaplasia were pooled (chromosome 10 loss, chromosome 9p loss, EGFR amplification, p16 deletion, PTEN mutation and telomerase reactivation), nongrowing tumours showed significantly fewer alterations than growing tumours (P=0.029). Among the whole series, no alteration could be correlated to a nontumorigenic feature.
Genetic alterations after establishment in nude mice
Comparison of genetic alterations was made between primary tumours (at the time of surgery) and first transplantations in nude mice, and then between the first and subsequent transplantations. Tumours showed a striking genetic stability before and after establishment in mice (Table 2). No alteration disappeared after xenograft and only two new alterations appeared in xenografts (a 1p loss for ODA-4-GEN and a PTEN homozygous deletion for ODA-20-THO). After the first transplantation, the molecular profile of alterations remained constant.
Correlations between spontaneous growth rate and genetic alterations
The latency (time between the first transplantation and the appearance of a palpable tumour) and the doubling time for 12 growing tumours are summarised in Table 3 showing that even if latency differed between tumours, the doubling time remained relatively stable. For the different genetic alterations tested, multivariate analysis showed that only EGFR amplification was associated with a higher growth rate (P=0.0082).
Discussion
These data indicate that human xenografts are useful models to study the molecular pathways involved in malignant oligodendrogliomas and astrocytomas. However, successful grafting occurred only in anaplastic tumours in terms of pathological grading of the original tumours, genotype analysis and prognostic significance for donor patients.
None of the grafted low-grade gliomas grew on nude mice while (45%) 23/51 high-grade gliomas did. Even among malignant gliomas, there was a significant difference, suggesting that higher grade tumours were preferentially selected by the grafting process since 62% (13/21) GBM or GBMO (grade IV) grew as compared to 33% (10/30) AO, AOA (grade III) (P=0.0047).
The results of the molecular analysis of the primary tumour (before grafting) are in agreement with the pathological data. Overall, growing tumours had significantly more molecular alterations than the nongrowing ones. This finding is particularly striking for EGFR amplification and LOH on chromosome 10, two alterations highly associated with increased malignancy (Bigner and Vogelstein, 1990; von Deimling et al, 1992; Reifenberger et al, 1996). It has previously been shown that EGFR amplification gives a tumorigenic advantage in GBM (Humphrey et al, 1988; Muleris et al, 1994), a feature that we often found associated with stable double-minute extra-chromosomal elements on caryotypic analysis (data not shown) (Bigner et al, 1989). In addition, EGFR amplification was present in 4/11 successfully grafted oligodendrogliomas in our series, indicating that the tumorigenic role of this alteration affects various subtypes of gliomas. Interestingly, overexpression of EGFR in transgenic mice has been shown to be involved in tumour progression of oligodendroglioma (Ding et al, 2003).
Similarly, we found that LOH on chromosome 10 was also extremely frequent not only in GBM (100%, 7 out of 7) but also in growing oligodendrogliomas (73%, eight out of 11). Thus, the loss of the putative tumour suppressor gene located on chromosome 10 is important for tumorigenicity in both tumour subtypes. However, we did not find evidence of involvement of the PTEN gene, which is located on chromosome10, in this series.
The fact that pathological and molecular factors predictive of successful grafting were associated with higher malignancy explaining that survival was shorter in donor patients whose tumours grew in nude mice (9.1 months) as compared to survival of patients whose tumours were rejected (15.4 months) (P=0.0003).
An important characteristic of this model is the stability of the genetic alterations in the xenografts as compared to the primary tumours, a finding also observed after successive passages in nude mice. Such stability, which was somewhat controversial in previous studies (Bigner et al, 1989; Muleris et al, 1994; Goike et al, 1999; Jeuken et al, 2000), appears to hold both for GBM and anaplastic oligodendrogliomas. Nevertheless, rare differences before and after growth on nude mice were shown as illustrated in Table 2. Rather than selection of a minor subclone, it is likely, as shown in Figure 2, that these alterations were already present in the original tumour, but were hardly visible or even undetectable because of the presence of ‘contaminating’ normal tissue.

Loss of heterozygosity patterns before and after establishment on nude mice. Genescan patterns showed a hardly detectable LOH in the initial tumour, which in contrast appeared very clearly after the first passage onto nude mice.
An advantage of this model is therefore to obtain tumours devoid of normal cells, improving the quality of molecular analyses (Figure 2).
Despite a stable genetic profile, the phenotype of the grafted tumour cells underwent striking changes whatever the histological features of the original tumour. After xenograft, all but one glioma lost their glial differentiation and presented a dedifferentiated phenotype in contrast with xenografts derived from other human cancers that maintained several of their histological characteristics (Rofstad et al, 1990a, 1990b; Poupon et al, 1993; Kolfschoten et al, 2000; Bras-Goncalves et al, 2001; de Pinieux et al, 2001; Krasagakis et al, 2001).
Whether the morphological homogeneity of grafted tumours represents a nonspecific change or supports the view that many gliomas have a common cell of origin (Daumas-Duport et al, 1997) leading to various morphological appearance in patients remains unsettled.
In summary, xenografting malignant astrocytomas and oligodendrogliomas is a useful method for studying and probably refining knowledge of the spectrum of genetic alterations in these tumours. This method can be of help in analysing the correlations between genotype and response to chemotherapy or various experimental agents. In addition, it may further benefit from new approaches such as gene expression by microarray studies in order to identify genes associated with chemosensitivity.
Change history
16 November 2011
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Bigner SH, Schold SC, Friedman HS, Mark J, Bigner DD (1989) Chromosomal composition of malignant human gliomas through serial subcutaneous transplantation in athymic mice. Cancer Genet Cytogenet 40: 111–120
Bigner SH, Vogelstein B (1990) Cytogenetics and molecular genetics of malignant gliomas and medulloblastoma. Brain Pathol 1: 12–18
Bras-Goncalves RA, Pocard M, Formento JL, Poirson-Bichat F, De Pinieux G, Pandrea I, Arvelo F, Ronco G, Villa P, Coquelle A, Milano G, Lesuffleur T, Dutrillaux B, Poupon MF (2001) Synergistic efficacy of 3n-butyrate and 5-fluorouracil in human colorectal cancer xenografts via modulation of DNA synthesis. Gastroenterology 120: 874–888
Cairncross JG, Ueki K, Zlatescu MC, Lisle DK, Finkelstein DM, Hammond RR, Silver JS, Stark PC, Macdonald DR, Ino Y, Ramsay DA, Louis DN (1998) Specific genetic predictors of chemotherapeutic response and survival in patients with anaplastic oligodendrogliomas. J Natl Cancer Inst 90: 1473–1479
Daumas-Duport C, Tucker ML, Kolles H, Cervera P, Beuvon F, Varlet P, Udo N, Koziak M, Chodkiewicz JP (1997) Oligodendrogliomas. Part II: a new grading system based on morphological and imaging criteria. J Neurooncol 34: 61–78
de Pinieux G, Legrier ME, Poirson-Bichat F, Courty Y, Bras-Goncalves R, Dutrillaux AM, Nemati F, Oudard S, Lidereau R, Broqua P, Junien JL, Dutrillaux B, Poupon MF (2001) Clinical and experimental progression of a new model of human prostate cancer and therapeutic approach. Am J Pathol 159: 753–764
Ding H, Shannon P, Lau N, Wu X, Roncari L, Baldwin RL, Takebayashi H, Nagy A, Gutmann DH, Guha A (2003) Oligodendrogliomas result from the expression of an activated mutant epidermal growth factor receptor in a RAS transgenic mouse astrocytoma model. Cancer Res 63: 1106–1113
European Agency for the Evaluation of Medicinal Products. Committee for Proprietary Medicinal Products (1999) Note for guidance on the pre-clinical evaluation of anticancer medicinal products (www.eudra.org/emea)
EORTC Laboratory Research Division (2001) European Organization for Research and Treatment of Cancer (EORTC) Laboratory Research Division workshop on the role of in vivo pre-clinical models in the development of contemporary cancer therapeutics, Verona, Italy, 3 February 2001. Eur J Cancer 37: 1976–1980
Goike HM, Asplund AC, Pettersson EH, Liu L, Sanoudou D, Collins VP (1999) Acquired rearrangement of an amplified epidermal growth factor receptor (EGFR) gene in a human glioblastoma xenograft. J Neuropathol Exp Neurol 58: 697–701
Hamelin R, Laurent-Puig P, Olschwang S, Jego N, Asselain B, Remvikos Y, Girodet J, Salmon RJ, Thomas G (1994) Association of p53 mutations with short survival in colorectal cancer. Gastroenterology 106: 42–48
He J, Mokhtari K, Sanson M, Marie Y, Kujas M, Huguet S, Leuraud P, Capelle L, Delattre JY, Poirier J, Hoang-Xuan K (2001) Glioblastomas with an oligodendroglial component: a pathological and molecular study. J Neuropathol Exp Neurol 60: 863–871
Humphrey PA, Wong AJ, Vogelstein B, Friedman HS, Werner MH, Bigner DD, Bigner SH (1988) Amplification and expression of the epidermal growth factor receptor gene in human glioma xenografts. Cancer Res 48: 2231–2238
Hunter SB, Abbott K, Varma VA, Olson JJ, Barnett DW, James CD (1995) Reliability of differential PCR for the detection of EGFR and MDM2 gene amplification in DNA extracted from FFPE glioma tissue. J Neuropathol Exp Neurol 54: 57–64
Ino Y, Betensky RA, Zlatescu MC, Sasaki H, Macdonald DR, Stemmer-Rachamimov AO, Ramsay DA, Cairncross JG, Louis DN (2001) Molecular subtypes of anaplastic oligodendrogliomas: implications for patient management at diagnosis. Clin Cancer Res 7: 839–845
Jeuken JW, Sprenger SHE, Wesseling P, Bernsen AJJA, Suijkerbuijk RF, Roelofs F, Macville MVE, Gilhuis HJ, van Overbeeke JJ, Boerman RH (2000) Genetic reflection of glioblastoma biopsy material in xenografts: characterization of 11 glioblastoma xenograft lines by comparative genomic hybridization. J Neurosurg 92: 652–658
Kleihues P, Cavenee WK (eds) (2000) Tumors of the Central Nervous System; Pathology and Genetics: World Health Organization International Classification of Tumours. IARC Press, Lyon
Kolfschoten GM, Pinedo HM, Scheffer PG, Schluper HM, Erkelens CA, Boven E (2000) Development of a panel of 15 human ovarian cancer xenografts for drug screening and determination of the role of the glutathione detoxification system. Gynecol Oncol 76: 362–368
Krasagakis K, Almond-Roesler B, Geilen C, Fimmel S, Krengel S, Chatzaki E, Gravanis A, Orfanos CE (2001) Growth and characterization of a cell line from a human primary neuroendocrine carcinoma of the skin (Merkel cell carcinoma) in culture and as xenograft. J Cell Physiol 187: 386–391
Louis DN, Holland EC, Cairncross JG (2001) Glioma classification: A molecular reappraisal. Am J Pathol 159: 779–786
McLaurin J, Trudel GC, Shaw IT, Antel JP, Cashman NR (1995) A human glial hybrid cell line differentially expressing genes subserving oligodendrocyte and astrocyte phenotype. J Neurobiol 26: 283–293
Merrill JE, Matsushima K (1988) Production of and response to interleukin 1 by cloned human oligodendroglioma cell lines. J Biol Regul Homeost Agents 2: 77–86
Muleris M, Almeida A, Dutrillaux AM, Pruchon E, Vega F, Delattre JY, Poisson M, Malfoy B, Dutrillaux B (1994) Oncogene amplification in human gliomas: a molecular cytogenetic analysis. Oncogene 9: 2717–2722
Post GR, Dawson G (1992) Characterization of a cell line derived from a human oligodendroglioma. Mol Chem Neuropathol 16: 303–317
Poupon MF, Arvelo F, Goguel AF, Bourgeois Y, Jacrot M, Hanania N, Arriagada R, Le Chevalier T (1993) Response of small-cell lung cancer xenografts to chemotherapy: multidrug resistance and direct clinical correlates. J Natl Cancer Inst 85: 2023–2039
Reifenberger J, Ring GU, Gies U, Cobbers L, Oberstrass J, An HX, Niederacher D, Wechsler W, Reifenberger G (1996) Analysis of p53 mutation and epidermal growth factor receptor amplification in recurrent gliomas with malignant progression. J Neuropathol Exp Neurol 55: 822–831
Rofstad EK, Wahl A, Stokke T, Nesland JM (1990a) Establishment and characterization of six human melanoma xenograft lines. APMIS 98: 945–953
Rofstad EK, Zaffaroni N, Hystad ME (1990b) Heterogenous radiation and heat sensitivity in vitro of human melanoma xenograft lines established from different lesion in the same patient. Int J Radiat Biol 57: 1113–1122
Smith JS, Perry A, Borell TJ, Lee HK, O'Fallon J, Hosek SM, Kimmel D, Yates A, Burger PC, Scheithauer BW, Jenkins RB (2000) Alterations of chromosome arms 1p and 10q as predictors of survival in oligodendrogliomas, astrocytomas, and mixed oligoastrocytomas. J Clin Oncol 18: 636–645
von Deimling A, Louis DN, von Ammon K, Petersen I, Hoell T, Chung RY, Martuza RL, Schoenfeld DA, Yasargil MG, Wiestler OD, Seizinger BR (1992) Association of epidermal growth factor receptor gene amplification with loss of chromosome 10 in human glioblastoma multiforme. J Neurosurg 77: 295–301
Walker DG, Duan W, Popovic EA, Kaye AH, Tomlinson FH, Lavin M (1995) Homozygous deletions of the multiple tumor suppressor gene 1 in the progression of human astrocytomas. Cancer Res 55: 20–23
Workman P, Twentyman P, Balkwill F, Balmain A, Chaplin D, Double J, Embleton J, Newell D, Raymond R, S J, Stephens T, Wallace J (1998) United Kingdom Co-ordinating Committee on Cancer Research (UKCCCR) Guidelines for the welfare of animals in experimental neoplasia (Second edition). Br J Cancer 77: 1–10
Zhou XP, Li YJ, Hoang-Xuan K, Laurent-Puig P, Mokhtari K, Longy M, Sanson M, Delattre JY, Thomas G, Hamelin R (1999) Mutational analysis of the PTEN gene in gliomas: molecular and pathological correlations. Int J Cancer 84: 150–154
Acknowledgements
This work was supported by grants of the Fondation de France (N° 2002010913) and the Association pour la Recherche contre le Cancer (ARC 4700). PL was supported by grants of the ARC, the Fondation pour la Recherche Médicale (FRM) and the Association pour la Recherche sur les Tumeurs Cérébrales (ARTC). We are indebted to Dr J-P Lagarde for technical advice, and to V Bordier for housekeeping the mice.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Leuraud, P., Taillandier, L., Aguirre-Cruz, L. et al. Correlation between genetic alterations and growth of human malignant glioma xenografted in nude mice. Br J Cancer 89, 2327–2332 (2003). https://doi.org/10.1038/sj.bjc.6601466
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.bjc.6601466
Keywords
This article is cited by
-
Glioblastoma-derived spheroid cultures as an experimental model for analysis of EGFR anomalies
Journal of Neuro-Oncology (2011)
