
Abstract
Differential microsatellite instability (MSI) in tumour epithelial and stromal compartments has not been well examined for colorectal cancers. Using laser-captured microdissection, separate specimens of these compartments of 40 sporadic colorectal cancers were sampled and MSI was tested with four markers. To examine the relation between the MSI phenotype in the stroma and other genetic events and histopathological features, p53 and K-ras gene mutations were analysed, and the expression of p53, hMLH1, and hMSH2 protein was determined by immunohistochemistry. Microsatellite instability positive results were obtained for both epithelium (34%) and stromal tissue (41%). While MSI in epithelium correlated with differentiation and Dukes' stage, that in stroma demonstrated an inverse relation, being particularly frequent in well-differentiated adenocarcinomas (54%) and Dukes' A lesions (55%). Further, a significant inverse correlation between p53 protein overexpression in the epithelium and MSI in the stroma was found (P=0.02475). The results suggest an alternative pathway of carcinogenesis involving stromal genetic instability in the development of colorectal cancers.
Similar content being viewed by others

Main
Interactions between epithelial and mesenchymal cells in the various organs play important roles in their development (Marsh and Trier, 1974; Shekhar et al, 2001), differentiation (Camps et al, 1990; Hom et al, 1998) and growth (Camps et al, 1990; Hom et al, 1998). However, the contribution of stromal cells to the generation and progression of epithelial neoplasia has not been thoroughly investigated. Although genetic alterations are frequently observed in many benign and malignant epithelial tumours in the form of microsatellite instability (MSI) (Michael-Robinson et al, 2001; Ward et al, 2001), the possibility of genetic abnormalities in the background microenvironment of these tumours has generally not been addressed properly. However, recent findings indicate that loss of heterozygosity (LOH) may be frequent in mammary stromal tissue in breast cancer patients (Moinfar et al, 2000). In one series of sporadic colorectal cancers, it was found that approximately 15% of tumours demonstrated MSI (Michael-Robinson et al, 2001). Since data on MSI in stroma of colorectum have hitherto not been published, to our knowledge, here we investigated this point with reference to clinicopathological features of colorectal cancers, using laser-captured microdissection. A recent study showed high p53 gene mutation loads in ulcerative colitis with an inflammatory microenvironment predisposed to colorectal carcinoma (Hussain et al, 2000; Yoshida et al, 2003). To test the hypothesis that genetic instability in the p53 locus stage might be a key early event in tumorigenesis, with change in the stroma possible influencing epithelial tumorigenesis, microsatellite markers D17S796 (Gyapay et al, 1994), TP53 and D17S786 (Gyapay et al, 1994), located in the short arm of chromosome 17 within 4 cM proximity to the p53 gene (17p13), and D17S579 (17q21) (Anderson et al, 1993) in the long arm of the chromosome 17 were analysed here in a series of sporadic colorectal cancers.
Materials and methods
Samples and preparation
In total, 40 surgically resected sporadic colorectal adenocarcinomas from patients, 24–89-years old, undergoing treatment at Kitasato University Hospital and Kitasato University East Hospital, were randomly selected. Histological typing was performed according to the criteria of the Japanese Society for Cancer of the Colon and Rectum (Jinnai, 1983) and also Dukes' classification.
On surgical removal, tissues were immediately frozen with liquid nitrogen in OCT compound for storage at −80°C. Frozen sections 10 (10 μm thick) were fixed in 70% alcohol and stained with Mayer's haematoxylin. Neoplastic epithelial and adjacent stromal tissues in the lamina propria were carefully microdissected with a laser-captured microdissection system (LM200, Arcturus, Mountain View, CA, USA) to avoid contamination (Figure 1A). Tissues from whole sections, including both epithelial and stromal elements (mixed), were also manually dissected. Tumour tissues with strong inflammatory cell infiltration were excluded from study. Likewise, normal mucosa from the colorectum of the same patients was sampled and processed as an internal control. Tissues were lysed in sodium dodecyl sulphate-lysis buffer with proteinase K, and DNA was extracted with the standard phenol–chloroform–ethanol precipitation method.

(A) Example of microdissection of epithelial cells and adjacent stromal cells from a single histologic section. Original magnification, × 100, (B) Representive data from microsatellite analysis of sporadic colorectal cancers. Arrows indicate shifted bands reflecting microsatellite instability. N=normal mucosa; T=tumour tissue.
Microsatellite analysis
The polymerase chain reaction (PCR) was performed for four microsatellite markers, D17S796, TP53 (forward: 5′-ACTGCCACTCCTTGCCCCATTC-3′, reverse: 5′-CACCTCGGGCTGAATAGTATCCCT-3′), D17S786, and D17S579, selected for analysing allelic instability in chromosome 17. Microdissected DNA (5–10 ng) was amplified with a Rapid Cycler (Idaho Technology, Idaho falls, ID, USA) and Takara Ex Taq DNA polymerase (Takara, Kyoto, Japan) under conditions as follows: 94°C for 3 min as the initial step, 35 cycles of 0 s at 94°C, 0 s at an appropriate temperature for each marker amplification, 6 s at 74°C, and a final step of 74°C for 3 min. The PCR products were fractionated by 3.6% polyacrylamide gel electrophoresis, fixed with 10% formamide and visualised using a Silver Stain Plus Kit (Bio-Rad, Hercules, CA, USA).
Results for normal tissues were considered informative when two or three (because some samples were insufficient for PCR amplification even if the experiment was repeated) of three samples, including epithelium, stroma, and mixed tissue DNA from each case, exhibited identical banding patterns. In pairs with informative normal tissue, sufficient PCR amplification of tumour tissues was considered as informative and MSI was defined as either a marked alteration in repeat length or as a new discrete band above or below the expected allele (Figure 1B). We referred to a previous report to judge whether the samples were informative (Luttges et al, 2001). Cases for which pairs of samples were informative for two of four markers were defined as informative and those showing MSI positivity (MSI+) for at least one marker were considered as MSI+.
Immunohistochemistry and p53 and K-ras gene analysis
To analyse p53, hMLH1, and hMSH2 protein expression and for assessment of p53 and K-ras gene mutations, tissues were fixed routinely in 10% buffered formalin and embedded in paraffin. Serial sections (3 μm thick) were applied for haematoxylin and eosin staining, immunohistochemistry, and mutation analyses.
Immunohistochemical staining was performed with monoclonal anti-p53 (DO7, × 300 dilution, Novocastras Lab., Newcastle, UK), monoclonal anti-hMLH1 (Clone; G168-15, × 200 dilution, BD PharMingen, San Diego, CA, USA) and monoclonal anti-hMSH2 (Clone; G219-1129, × 500 dilution, BD PharMingen) antibodies, using the standard labelled streptavidin–biotin–peroxidase complex method described in our previous report (Yamashita et al, 2001). The amounts of positive cells were expressed as the percentage of the total number of epithelial cells and assigned to one of three categories for p53: ++, > 50%; +, 0.5–50%; −, < 0.5% (Figure 3I–K), for hMLH1 and hMSH2: ++, > 50%; +, 10–50%; −, < 10% (Figure 3A–F). We also examined the expression of hMLH1 and hMSH2 proteins in stromal cells, with classification into: ++, strong staining; +, focal staining; −, negative staining. Definite nuclear staining of adjacent non-neoplastic epithelial and stroma cells or lymphocytes served as internal positive controls.

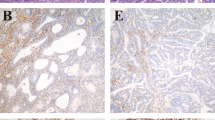
Expression of hMLH1, hMSH2, and p53 protein. Examples of hMLH1 (A–C) and hMSH2 (D–F) protein immunostaining in tumorous epithelial cells. (A) and (D) − in epithelial cells; + in stromal cells (B) and (E) + in epithelial cells; + in stromal cells. (C) and (F) ++ in epithelial cells; ++ in stromal cells. Original magnification, × 200. (G) hMLH1 protein expression in normal stromal cells. (H) Loss of hMLH1 protein expression in tumorous stromal cells from the same case. Original magnfication, × 400. Examples of p53 protein immunostaining in sporadic colorectal cancers (I–K). (I) − for p53 protein expression; (J) +; (K) ++. Original magnification, × 400.
p53 and K-ras gene mutations were analysed by the PCR-single strand conformation polymorphism (SSCP) method described in our previous report (Yamashita et al, 2001). Mutations were detected as abnormally shifted bands.
Statistics
Differences for each category of clinicopathological features, with reference to MSI in the epithelium and stroma were examined using the Fisher' s exact and χ2 tests.
Results
MSI status
Microsatellite instability was frequently detected in epithelial and stromal areas of sporadic colorectal cancers (Table 1). Of the 40 colorectal cancers studied, 13 out of 38 informative cases (34%) were MSI+ for one or more of the markers in tumour epithelium, 16 out of 39 cases (41%) in adjacent stromal areas, and nine of 40 (23%) in mixed tissue. MSI+ for two or more markers was found in three cases in the epithelium and two in stroma. While MSI was slightly more common in the stroma than epithelium, the difference did not reach significance (Table 1). For each component, only three cases had MSI in both epithelium and stroma for the same markers (two for TP53 and one for D17S796), suggesting appropriate microdissection without contamination. Others showed MSI specific to the epithelium alone, stroma alone, epithelium and mixed tissue (Figure 1B; D17S786), or stroma and mixed tissue. All the tumours with MSI in mixed tissue also demonstrated MSI in epithelium and stroma. A comparison of MSI frequency for each marker between the epithelium and stroma revealed stromal MSI+ to be less frequent (1/36=3%) than epithelial MSI+ (6/37=16%) for D17S786 (P=0.0512) (Table 1). With D17S796, MSI was more often found in the stroma (8/33=24%) than in the epithelium (4/36=11%), without significance (Table 1). No differences were found for D17S579 and TP53. Concerning the low MSI frequency for mixed tissue as compared with the epithelium or stroma alone, MSI−DNA might disturb the positivity with MSI-PCR due to lowered sensitivity.
Histopathological features of epithelial and stromal MSI status
Histopathological and molecular features of the sporadic colorectal cancers are detailed in Table 2. MSI frequencies differed between epithelium and stroma in well-differentiated (P-value=0.0393) and poorly differentiated (P-value=0.0510) (Figure 2A) adenocarcinomas. Stromal MSI+ was more often detected in well-differentiated adenocarcinomas (7/13=54%) than in poorly differentiated cancers (1/10=10%) (P-value=0.0286), whereas the frequency of epithelial MSI+ correlated with progression (Figure 2A). With analysis of Dukes' stage, although there was a tendency for an inverse relation with stromal MSI+ (Figure 2B), it did not reach significance (P-value=0.4807). In contrast, epithelial MSI+ showed significant variation with the Dukes' stage (P-value=0.0277) (Figure 2B). A significant correlation was also detected between epithelial MSI+ and stromal MSI+ and Dukes' stage (P-value=0.0455). It is notable that in stage A lesions, stromal MSI+ (6/11=55%) was more frequent than epithelial MSI+ (0/10=0%) (P-value=0.0057) (Figure 2B).

Data for MSI with reference to tumour features. E=epithelium; S=stroma; M=mixed tissue. *P<0.01; #P<0.05.
MSI status, p53 gene mutations, and p53 protein overexpression
A significant inverse correlation between p53 protein overexpression in epithelium and MSI in stroma was found (7/10=70% in ‘+’ cases vs 6/22=27% in ‘++’ cases) (P-value=0.02475) (Figure 2C). p53 gene mutations were detected in 15 out of 40 (38%) tumours with no significant differences observed with reference to epithelial and stromal MSI (Table 2).
MSI status and K-ras gene mutation
K-ras gene mutations were detected in 12 out of 40 (30%) tumours with no significant differences observed with reference to epithelial and stromal MSI (Table 2).
hMLH1 and hMSH2 protein expression in epithelial and stromal MSI
Losses of hMLH1 and hMSH2 expression were detected in 10% (4/40) and 3% (1/40) of cases in the epithelium and 3% (1/40) and 0% (0/40) in the stroma, respectively. No significant correlations between hMLH1 and hMSH2 protein expression in epithelial or stromal cells and epithelial MSI+ or stromal MSI+ were found (Table 2) (P-value not shown).
Discussion
Regarding interactions between the epithelium and stroma, several hypotheses have been proposed to explain fibroblast-promoting effects on tumour growth. Most of the intercellular material, the extracellular matrix (ECM) molecules that are required for tumour growth and progression, is produced by stromal cells (Noel et al, 1998). It has been demonstrated that neoplastic breast stroma drives alteration in gene expression as compared with normal tissue (Leygue et al, 1998). In fact, it is generally believed that the epithelium is the neoplastic element in most tumours and that altered gene expression in stroma occurs as the secondary reaction. However, the recent finding of frequent genetic changes in mammary stromal tissue in breast cancer patients (Moinfar et al, 2000), and the demonstration that inflammation-associated stroma promotes conversion of colonic adenoma cells to adenocarcinoma cells in nude mice (Okada et al, 2000) suggest a more complex scenario.
Our present study showed that MSI in stromal and epithelial elements can occur independently in sporadic colorectal cancers, in line with the previous findings for breast carcinomas (Kurose et al, 2001). Further, while MSI in the epithelium tended to correlate with differentiation and the Dukes' stage, the inverse was the case for MSI in stroma. These interesting results strongly suggest that there are alternative mechanisms involving stromal MSI operating in colorectal carcinogenesis and progression. According to Young et al, methylation of CpG island occurs both in the epithelium and stroma (Young et al, 2001). Stromal MSI presented in this study might be due to the methylation of mismatch repair enzymes in stromal cells, although the identification of the enzyme remains unclear. Previously, it was shown that high-level MSI (MSI-H) tumours are more likely to be right sided than their low-level MSI (MSI-L) or MSI stable counterparts (Michael-Robinson et al, 2001; Ward et al, 2001). In the present study, while MSI frequencies in both the epithelium and stroma were high in right-side (43, 50%) as compared to left-side lesions (29, 36%), the difference did not reach statistical significance. This might be due to relatively small numbers of examined cases or inclusion of both MSI-L and MSI-H results in our analysis. Additionally, we examined the expression of DNA mismatch repair enzymes, hMLH1 and hMSH2 protein, in epithelial and stromal cells, respectively, as the MSI-H phenotype has been suggested to be of importance for the DNA mismatch repair system in sporadic colorectal cancers (Dietmaier et al, 1997; Ward et al, 2001). In the present study, epithelial MSI+ for two or more markers was found in three cases, two of which showed loss of hMLH1 protein expression. In stroma, MSI+ for two or more markers was found in two cases, one of which showed loss of MLH1 in stromal cells (Figure 3G and H). Although we have not used the standard markers that were recommended for MSI analysis on the basis of a National Cancer Institute Workshop (Boland et al, 1998), the results of hMLH1 and hMSH2 protein expression provide support for the validity of our study of MSI in epithelial and stromal cells. NCI-recommended standard markers are two mononucleotide repeat markers and three dinucleotide markers, but all of the markers we used in this study are dinucleotide repeat markers ((CA)n repeat). Therefore, it is difficult to compare MSI-H entity with our MSI+ for two or more markers. Additionally, discordance of the loss of hMLH1 or hMSH2 and our MSI+ tumours could occur in the present study.
Although no significant links were found between MSI and p53 or K-ras gene mutations, MSI in the epithelium, but not stroma, tended to correlate positively with p53 protein overexpression. Recently, Kapiteijn et al. (2001) reported that p53 gene mutation corresponds more often to p53 overexpression in left- than in right-sided tumours, suggesting that mechanisms of oncogenesis may differ between the two cases. This is in line with our results for a greater prevalence of p53 overexpression with p53 gene mutations in left-sided tumours (9/11, 81%), than in those found on the right side (1/4, 25%) (P=0.0390, data not shown). Furthermore, we found a difference in the relation between p53 mutations and stromal MSI (stromal MSI+/p53 mutation, 3/11 (27%) in the left-side; 3/4 (75%) in the right-side, P=0.0952). These results support the conclusion of Kapiteijn et al. (2001) and suggest a relation of p53 mutation to stromal MSI+.
From the available data, contrary to the general belief that abnormalities in stroma occur as reactions to epithelial tumour cells, we propose the hypothesis that an alternative pathway may exist, with stromal genetic instability influencing epithelial cells in carcinogenesis.
Change history
16 November 2011
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Anderson LA, Friedman L, Osborne-Lawrence S, Lynch E, Weissenbach J, Bowcock A, King MC (1993) High-density genetic map of the BRCA1 region of chromosome 17q12–q21. Genomics 17: 618–623
Boland CR, Thibodeau SN, Hamilton SR, Sidransky D, Eshleman JR, Burt RW, Meltzer SJ, Rodriguez-Bigas MA, Fodde R, Ranzani GN, Srivastava S (1998) A National Cancer Institute Workshop on Microsatellite Instability for cancer detection and familial predisposition: development of international criteria for the determination of microsatellite instability in colorectal cancer. Cancer Res 58: 5248–5257
Camps JL, Chang SM, Hsu TC, Freeman MR, Hong SJ, Zhau HE, von Eschen-bach AC, Chung LW (1990) Fibroblast-mediated acceleration of human epithelial tumour growth in vivo. Proc Natl Acad Sci USA 87: 75–79
Dietmaier W, Wallinger S, Bocker T, Kullmann F, Fishel R, Ruschoff J (1997) Diagnostic microsatellite instability: definition and correlation with mismatch repair protein expression. Cancer Res 57: 4749–4756
Gyapay G, Morissette J, Vignal A, Dib C, Fizames C, Millasseau P, Marc S, Bernardi, G, Lathrop M, Weissenbach J (1994) The 1993–94 Genethon human genetic linkage map. Nat Genet 7: 246–339
Hom YK, Young P, Wiesen JF, Miettinen PJ, Derynck R, Werb Z, Cunha GR (1998) Uterine and vaginal organ growth requires epidermal growth factor receptor signaling from stroma. Endocrinology 139: 913–921
Hussain SP, Amstad P, Raja K, Ambs S, Nagashima M, Bennett WP, Shields PG, Ham AJ, Swenberg JA, Marrogi AJ, Harris CC (2000) Increased p53 mutation load in noncancerous colon tissue from ulcerative colitis: a cancer-prone chronic inflammatory disease. Cancer Res 60: 3333–3337
Jinnai D (1983) General rules for clinical and pathological studies on cancer of the colon, rectum and anus. Part I. Clinical classification. Japanese Research Society for Cancer of the Colon and Rectum. Jpn J Surg 13: 557–573
Kapiteijn E, Liefers GJ, Los LC, Kranenbarg EK, Hermans J, Tollenaar R.A., Moriya Y, van de Velde CJ, van Krieken JH (2001) Mechanisms of oncogenesis in colon versus rectal cancer. J Pathol 195: 171–178
Kurose K, Hoshaw-Woodard S, Adeyinka A, Lemeshow S, Watson PH, Eng C (2001) Genetic model of multi-step breast carcinogenesis involving the epithelium and stroma: clues to tumour-microenvironment interactions. Hum Mol Genet 10: 1907–1013
Leygue E, Snell L, Dotzlaw H, Hole K, Hiller-Hitchcock T, Roughley PJ, Watson PH, Murphy LC (1998) Expression of lumican in human breast carcinoma. Cancer Res 58: 1348–1352
Luttges J, Galehdari H, Brocker V, Schwarte-Waldhoff I, Henne-Bruns D, Kloppel G, Schmiegel W, Hahn SA (2001) Allelic loss is often the first hit in the biallelic inactivation of the p53 and DPC4 genes during pancreatic carcinogenesis. Am J Pathol 158: 1677–1683
Marsh MN, Trier JS (1974) Morphology and cell proliferation of subepithelial fibroblasts in adult mouse jejunum. II. Radioautographic studies. Gastroenterology 67: 636–645
Michael-Robinson JM, Biemer-Huttmann A, Purdie DM, Walsh MD, Simms LA, Biden KG, Young JP, Leggett BA, Jass JR, Radford-Smith GL (2001) Tumour infiltrating lymphocytes and apoptosis are independent features in colorectal cancer stratified according to microsatellite instability status. Gut 48: 360–366
Moinfar F, Man YG, Arnould L, Bratthauer GL, Ratschek M, Tavassoli FA (2000) Concurrent and independent genetic alterations in the stromal and epithelial cells of mammary carcinoma: implications for tumorigenesis. Cancer Res 60: 2562–2566
Noel A, Hajitou A, L'Hoir C, Maquoi E, Baramova E, Lewalle JM, Remacle A, Kebers F, Brown P, Calberg-Bacq CM, Foidart JM (1998) Inhibition of stromal matrix metalloproteases: effects on breast-tumor promotion by fibroblasts. Int J Cancer 76: 267–273
Okada F, Kawaguchi T, Habelhah H, Kobayashi T, Tazawa H, Takeichi N, Kitagawa T, Hosokawa M (2000) Conversion of human colonic adenoma cells to adenocarcinoma cells through inflammation in nude mice. Lab Invest 80: 1617–1628
Shekhar MP, Werdell J, Santner SJ, Pauley RJ, Tait L (2001) Breast stroma plays a dominant regulatory role in breast epithelial growth and differentiation: implications for tumor development and progression. Cancer Res 61: 1320–1326
Ward R, Meagher A, Tomlinson I, O'Connor T, Norrie M, Wu R, Hawkins N (2001) Microsatellite instability and the clinicopathological features of sporadic colorectal cancer. Gut 48: 821–829
Yamashita K, Yoshida T, Shinoda H, Okayasu I (2001) Novel method for simultaneous analysis of p53 and K-ras mutations and p53 protein expression in single histologic sections. Arch Pathol Lab Med 125: 347–352
Yoshida T, Mikami T, Mitomi H, Okayasu I (2003) Diverse p53 alterations in ulcerative colitis-associated low-grade dysplasia: full-length gene sequencing in microdissected single crypts. J Pathol 199: 166–175
Young J, Biden KG, Simms LA, Huggard P, Karamatic R, Eyre HJ, Sutherland GR, Herath N, Barker M, Anderson GJ, Fitzpatrick DR, Ramm GA, Jass JR, Leggett BA (2001) HPP1: a transmembrane protein-encoding gene commonly methylated in colorectal polyps and cancers. Proc Natl Acad Sci USA 98: 265–270
Acknowledgements
This work has been supported by Grants-in-Aid for Scientific Research from the Japanese Ministry of Education, Culture, Sports, Science and Technology (no. 13214093), and Scientific Research from Kitasato University Postgraduate School of Medical Sciences (no. 4002).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Matsumoto, N., Yoshida, T., Yamashita, K. et al. Possible alternative carcinogenesis pathway featuring microsatellite instability in colorectal cancer stroma. Br J Cancer 89, 707–712 (2003). https://doi.org/10.1038/sj.bjc.6601141
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.bjc.6601141
Keywords
This article is cited by
-
Genetic instability in the tumor microenvironment: a new look at an old neighbor
Molecular Cancer (2015)
-
Unravelling mechanisms of p53-mediated tumour suppression
Nature Reviews Cancer (2014)
-
p53 regulates mesenchymal stem cell-mediated tumor suppression in a tumor microenvironment through immune modulation
Oncogene (2014)
-
Role of Stromal-Epithelial Interaction in the Formation and Development of Cancer Cells
Cancer Microenvironment (2013)
-
Cervical carcinoma-associated fibroblasts are DNA diploid and do not show evidence for somatic genetic alterations
Cellular Oncology (2011)
