
Abstract
Intracellular redistribution of β-catenin through mutation of the adenomatous polyposis coli (APC) gene has been proposed as an early tumorigenic event in most colorectal tumours. In serrated adenoma (SA), a newly recognised subtype of colorectal adenoma, APC mutations are uncommon, and the contribution of β-catenin to tumorigenesis remains unclear. We compared intracellular localisation of β-catenin and presence of mutations in exon 3 of β-catenin between 45 SAs, with 71 conventional adenomas (CADs), and eight carcinomas invading the submucosa (SCAs). Widespread or focal nuclear β-catenin expression was demonstrated in 7% of SAs (three out of 45), 61% of CADs (43 out of 71), and 88% of SCAs (seven out of eight). Cytoplasmic immunostaining for β-catenin was demonstrated in 16% of SAs (seven out of 45), 77% of CADs (55 out of 71), and 88% of SCAs (seven out of eight). No mutation in exon 3 of β-catenin was found in SAs or SCAs, while 7% of CADs (five out of 71) had β-catenin mutations. No nuclear or cytoplasmic expression of β-catenin was observed in the hyperplastic or conventionally adenomatous epithelium of mixed-type SAs. These findings suggest that β-catenin mutation is unlikely to contribute to the tumorigenesis in SA, and that intracellular localisation of β-catenin may not be associated with an early event of the tumour progression in most SAs.
Similar content being viewed by others

Main
The adenoma–carcinoma sequence of colorectal carcinogenesis requires an accumulation of genetic alterations. Inactivation of the adenomatous polyposis coli (APC) suppressor gene is an initial event in the development of most colorectal tumours (Kinzler and Vogelstein, 1996). This inactivation leads to a decrease in APC-dependent degradation of the oncoprotein β-catenin, thereby increasing its cytoplasmic abundance (Korinek et al, 1997; Morin et al, 1997). The β-catenin protein is involved in two functions: cell-to-cell adhesion and mediation of the Wingless/Wnt signal transduction pathway (Behrens et al, 1996; Barth et al, 1997); the latter action is associated with the oncogenic potential of this protein. This pathway involves stabilisation of the cytoplasmic pool of β-catenin, translocation of β-catenin to the nucleus, complex formation between β-catenin and transcription factors of the T-cell factor (Tcf) family, and activation of this transcriptional complex resulting in constitutive transcriptional activation of downstream target genes that regulate cell proliferation or apoptosis (Korinek et al, 1997; Morin et al, 1997). Recently, c-myc (He et al, 1998) and cyclin D1 (Tetsu and McCormick, 1999) have been identified as target genes of the β-catenin/Tcf complex. Such signal transduction involving β-catenin plays a critical role in colorectal carcinogenesis.
Recently, mutation in exon 3 of β-catenin has been identified in approximately half of colorectal tumours that lack APC mutations (Morin et al, 1997). These β-catenin mutations involve alterations of exon 3 of serine/threonine sites that normally are phosphorylated by glycogen synthatase kinese 3β (GSK-3β). As phosphorylation of these sites is apparently necessary for APC-induced degradation of β-catenin (Morin et al, 1997), the β-catenin mutations cause intracellular β-catenin accumulation, increasing formation of the transcription-activating β-catenin/Tcf complex.
Some authors recently have reported that hyperplastic colorectal polyps may in fact be neoplastic lesions because they include some of the genetic alterations seen in colorectal cancers (CRC), such as Ki-ras mutations (Otori et al, 1997) and microsatellite instability (Jass et al, 2000b). Serrated adenoma (SA), which histologically shows hyperplastic architecture and epithelial dysplasia, has been recognised as a new subtype of colorectal adenoma (Longacre and Fenoglio-Preiser, 1990). Given an incidence of high-grade dysplasia in SAs of 10–14.8% (Longacre and Fenoglio-Preiser, 1990; Matsumoto et al, 1999), SAs have been considered potential precursors of colorectal carcinoma. Some reports have concluded that colorectal carcinoma may be associated with SA (Hiyama et al, 1998; Makinen et al, 2001). On the other hand, SAs often occur in contiguity with typical hyperplastic polyps (Iino et al, 1999). While some investigators have suggested that genetic alterations in SAs differ from those in conventional adenomas (CADs) (Ajioka et al, 1998; Hiyama et al, 1998; Uchida et al, 1998; Iino et al, 1999; Jass et al, 2000a; Rashid et al, 2000; Dehari, 2001; Hawkins and Ward, 2001), the differences have not been specified.
Recently, Dehari (2001) and Uchida et al (1998) have reported that APC gene mutation is uncommon in SA. However, the contribution of β-catenin to tumorigenesis in SA remains unclear. The aim of this study was to examine intracellular localisation of β-catenin and to identify any mutations in exon 3 of β-catenin in SAs, by comparing the findings with those in CADs and carcinomas invading the submucosa (SCAs).
Materials and methods
Subjects
A total of 45 SAs from 43 patients who underwent endoscopic (n=44) or transanal resection (n=1) at Showa University Hospital between September 1997 and December 2001 were analysed in this study. CADs (n=71) and SCAs (n=8) obtained endoscopically or surgically were studied for comparison. Informed consent was obtained from all patients. We excluded patients who had familial adenomatous polyposis, hereditary nonpolyposis colorectal cancer (HNPCC), or hyperplastic polyposis (Jass and Burt, 2000).
Histologic evaluation
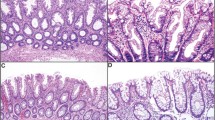
Specimens resected or obtained by endoscopy were fixed in 10% buffered formalin for 24 h, dehydrated, and embedded in paraffin. Serial sections (3 μm) were cut from the paraffin block to be prepared for haematoxylin and eosin (H&E) staining and immunostaining. All H&E-stained sections were reviewed by a senior pathologist (TY), who was blinded to colonoscopic findings. The histologic diagnosis of SA was based on the definition of Longacre and Fenoglio-Preiser (1990) (Figure 1). Histologic findings of (1) a serrated architecture simulating a hyperplastic polyp, (2) goblet cell immaturity, (3) mitotic figures in the upper crypt zone, (4) prominence of nuclei, and (5) cytoplasmic eosinophilia were the criteria for inclusion. SAs were subclassified as pure or mixed depending on whether or not a component other than SA was present. Epithelial dysplasia without serrated architecture was classified as CAD. When the tumour cells had spread through the muscularis mucosae into the submucosa, the lesion was defined as carcinoma. Clinicopathologic characteristics of patients with SAs, CADs, and SCAs are shown in Table 1. No significant difference was noted in the distribution for gender, age, location, size, or macroscopic type between SA and CAD groups.

Histologic appearance of a typical SA showing serrated architecture and epithelial dysplasia. H&E staining.
Immunohistochemical detection of β-catenin
Deparaffinised, rehydrated sections were heated in a microwave oven in 0.01 mol l−1 sodium citrate buffer (pH 6.0) for 15 min to retrieve antigens. Endogenous peroxidase activity was inhibited by incubation with 0.3% hydrogen peroxidase in methanol for 5 min. Sections were incubated with anti-β-catenin antibody (1 : 300, Transduction Labs, Lexington, KY, USA) for 90 min. Sections were then incubated with horseradish peroxidase (HRP)-binding amino-acid polymer for 30 min (Histofine Simplestain MAX-PO kit, Nichrei, Tokyo, Japan). Colour was developed by staining with diaminobenzidine solution. Sections were lightly counterstained with haematoxylin. Phosphate-buffered saline (pH 7.2) was used for rinsing between each step.
Evaluation of β-catenin immunostaining
Each immunostained section was examined under light microscopy and evaluated by a senior pathologist (TY) according to the modified scoring method of Hugh et al (1999) (Figure 2). Nuclear, cytoplasmic, and membranous stainings were considered separately. Nuclear expression by tumour cells was evaluated as widespread (>75% of cells per section), focal (<75% of cells per section), or absent (no nuclear staining). Cytoplasmic staining was evaluated as negative or positive. Staining intensity of the cell membrane was compared with that seen in normal mucosal cells of colorectum, and then was scored as: 2+ (preserved type), with tumour cells displaying well-localised membranous staining simulating normal mucosa; 1+ (reduced type), with tumour cells displaying reduced membranous immunoreactivity; 0 (absent type), with no immunostaining of tumour cells. Normal colonic epithelial cells were used as an internal control.

Immunohistochemical staining of β-catenin. (A) Widespread nuclear and cytoplasmic expression in a colonic adenoma with high-grade dysplasia. (B) Membrane expression of β-catenin in the normal mucosa adjacent to the colorectal neoplasms.
DNA preparation from pure-type SA, CAD, and SCA
To extract genomic DNA, five sections (each 5 μm thick) were obtained from an archival block of formalin-fixed, paraffin-embedded tumour tissue for each polyp type. One section was stained with H&E, and the percentage of tumour cells was estimated by microscopic examination. Representative tumour samples contained a minimum of 80% tumour cells. After deparaffinisation, DNA samples were extracted from the remaining four sections using standard proteinase K–phenol–chloroform methods.
Laser-capture microdissection for mixed-type SA
To exclude contamination between tumour types, separate samples were obtained from mixed-type SAs for adenomatous areas and other areas (i.e., hyperplastic tissue, conventionally adenomatous tissue, or carcinomatous tissue) using laser-capture microdissection. Deparaffinised sections were stained with H&E, followed by three dehydration steps of 60 s each in 70, 95, and 99.5% ethanol, with final dehydration in xylene. Once air-dried, the stained tissues were laser-capture microdissected by a PixCell II LCM system following the manufacturer's protocols (Acturus Engineering, Mountain View, CA, USA). After the tumour cells had been collected into a 0.5-ml reaction tube, DNA was isolated by adding 50 μl of proteinase K digestion buffer. The reaction tubes were incubated overnight at 37°C. After denaturation at 95°C for 8 min, the lysate was used for DNA analysis.
Detection of β-catenin-gene mutations
Exon 3 of β-catenin was examined for mutations using fluorescence-based polymerase chain reaction-single-strand conformation polymorphism (PCR-SSCP) analysis. Primers used for PCR were 5′-TGATTTGATGGAGTTGGACA-3′ (forward) and 5′-CTGTTCCCA-CTCATACAGGA-3′ (reverse). The PCR reaction was carried out in a 20 μl final volume, containing 50 ng DNA, 0.5 U Takara Ex Taq (Takara, Sigma, Japan), 1 μmol of each primer, 2.5 mM of each dNTP, and 10 × Ex Taq buffer (Takara). The following cycling protocol was used: 94°C for 1 min, followed by 40 cycles of 94°C for 30 s, 55°C for 1 min, and 72°C for 1 min, and then a final extension at 10°C for 60 min. PCR products were visualised and purified by electrophoresis on 2% agarose gels for 40 min at 100 V, and stained with ethidium bromide. These PCR products were used to detect β-catenin mutation by fluorescence-based SSCP analysis using an automated DNA sequencer (ALF Express; Amersham Pharmacia Biotech, Uppsala, Sweden), with an external cooling bath. Peak patterns were analysed using the ALFwin Fragment Analyser program (Amersham Pharmacia Biotech). Shifted peaks indicated the mutations in the DNA fragments. Nucleotide sequences of DNA fragments with shifted peaks were determined as described previously (Makino et al, 2000).
Statistical analysis
To determine statistical significance, relation between molecular and clinicopathologic data were analysed using χ2 test, or Fisher's exact test as appropriate. A value of P less than 0.05 was considered to indicate significance.
Results
Expression of β-catenin in normal colonic mucosa
Membranous staining of β-catenin uniformly localised at the intercellular borders was observed in the normal mucosa adjacent to colorectal neoplasms (Figure 2B). No nuclear or cytoplasmic immunostaining was seen in normal mucosa.
Nuclear expression of β-catenin
Table 2 shows nuclear expression of β-catenin in SAs, CADs, and SCAs. No widespread immunostaining was observed in the nuclei of SA cells, with nuclear expression being restricted to focal areas. Widespread or focal nuclear expression of β-catenin was demonstrated in only 7% of SAs (three out of 45), in contrast to 61% of CADs (43 out of 71) (P<0.0001). All but one SCA specimen was positive for nuclear staining. In SAs and CADs, nuclear expression of β-catenin was observed more frequently in large (>10 mm) than in small (<10 mm) lesions. However, these size-related differences were not statistically significant.
Cytoplasmic and cell-membranous expression of β-catenin
Table 3 shows cytoplasmic expression of β-catenin in SAs, CADs, and SCAs. Cytoplasmic accumulation of β-catenin was demonstrated in 16% of SAs (seven out of 45) and 77% of CADs (55 out of 71: P<0.0001). All but one SCA specimen was positive for cytoplasmic staining. In CADs, cytoplasmic immunostaining for β-catenin was detected more frequently in large (⩾10 mm) than in small (<10 mm) CADs (P=0.0029). However, no significant difference was noted in cytoplasmic immunostaining of β-catenin between large and small SAs. No significant difference in cell-membrane expression of β-catenin could be found among SAs, CADs, and SCAs.
Detection of β-catenin mutation
No mutation in exon 3 of β-catenin was found in SAs or SCAs, while 7% of CADs (five out of 71) had β-catenin mutations. Considering the size of CADs, β-catenin mutations were detected in 8% of small CADs (<10 mm; three out of 39) and 6% of large CADs (>10 mm; two out of 32). Table 4 summarises tumour location, tumour size, substitution of one base, codon affected, and predicted amino-acid substitution. All mutations involved loss of serine or threonine residues from the GSK-3β phosphorylation region.
Immunohistochemical and genetic findings of β-catenin in mixed-type SAs
In the 21 mixed-type SAs, serrated and nonserrated areas were identified histologically as shown in Table 1. The 21 mixed-type SAs involved various nonserrated components as follows: 13 hyperplastic polyps, seven CAs, and one carcinoma that invaded the submucosal layer. No nuclear or cytoplasmic immunostaining for β-catenin was observed in the hyperplastic or conventionally adenomatous epithelium in the 20 mixed polyps. In the carcinomatous epithelium of mixed-type SAs, nuclear β-catenin-positive cells were focally detected, and cytoplasmic staining for β-catenin was observed (Figure 3). Neither nuclear nor cytoplasmic staining for β-catenin was found in the adjacent serrated epithelium. Microdissection allowed each area of mixed-type SA to be examined separately for a β-catenin mutation. However, no β-catenin mutation was found in serrated or nonserrated areas.

Immunohistochemical findings in a Dukes' A carcinoma associated with SA. The carcinoma invaded the submucosal layer. In (A), focal nuclear staining of cells for β-catenin is detected; and cytoplasmic staining for β-catenin is observed in the carcinomatous epithelium of this mixed-type SA. In (B), neither nuclear nor cytoplasmic staining for β-catenin is found in the adjacent serrated epithelium.
Discussion
Recent reports have described nuclear expression of β-catenin in familial and sporadic colorectal tumours (Inomata et al, 1996; Takayama et al, 1996; Hao et al, 1997; Valizadeh et al, 1997; Hugh et al, 1999; Kobayashi et al, 2000). Valizadeh et al (1997) have suggested that both cytoplasmic and nuclear localisation of β-catenin in dysplastic colonic polyps are early events in the development of colorectal cancer. Nuclear localisation has been linked to progression of colorectal carcinogenesis when a certain threshold tumour size is exceeded (Brabletz et al, 2000). However, Kobayashi et al (2000) have reported that nuclear translocation of β-catenin is involved in initiation of cancer in distinction to adenoma, independently of APC mutations. In our study, about 80% of CADs displayed either cytoplasmic or nuclear localisation of β-catenin, while nuclear overexpression was observed in 50% of SCAs (four out of eight). Abnormal distribution of β-catenin was detected more frequently in large CADs (⩾10 mm) than in smaller CADs. However, nuclear overexpression of β-catenin generally was not observed in our SAs; even focal nuclear staining was found in only 7% (three out of 45). Cytoplasmic accumulation of β-catenin was observed in 16% of SAs (seven out of 45). The nuclear and cytoplasmic localisation of β-catenin in SA thus differs from observations in CADs and SCAs.
We identified mutations in exon 3 of β-catenin in 7% of sporadic CADs (five out of 71). As in previous studies (Kitaeva et al, 1997; Morin et al, 1997; Sparks et al, 1998; Samowitz et al, 1999), all mutations involved the loss of serine or threonine residues from GSK-3β phosphorylation sites. Loss of these phosphorylation sites is thought to promote tumorigenesis through decreased APC-associated degradation of β-catenin and increased β-catenin/Tcf transcriptional activation (Morin et al, 1997).
One important reported finding in CADs is a difference in the incidence of β-catenin mutation between small and large CADs. Samowitz et al (1999) have suggested that adenomas with mutations in exon 3 of β-catenin are unlikely to progress to large adenomas or invasive cancers, since β-catenin mutations are significantly more common in very small adenomas than in large adenomas or invasive cancers. Mutations in β-catenin sometimes were seen in our large CADs as well as small CADs, with no significant difference in frequency according to lesion size (small, 6%; large, 8%). This disagreement with the previous study (Samowitz et al, 1999) suggests that mutation of β-catenin may be an early, nonsize-dependent event in the conventional adenoma–carcinoma sequence.
β-Catenin mutation occurs in primary CRC and CRC cell lines that lack APC mutation (Morin et al, 1997). APC mutation is infrequent in MSI-H cancers (Konishi et al, 1996; Salahshor et al, 1999). However, β-catenin mutation is not found in sporadic CRC with high level of microsatellite instability (MSI-H) (Salahshor et al, 1999). These observations fit with the lack of abnormal β-catenin immunostaining in sporadic MSI-H cancers (Wong et al, 2002). When β-catenin mutation is found in sporadic MSI-H cancers, the subjects are young and probably have HNPCC (Mirabelli-Primdahl et al, 1999). Moreover, Mirabelli-Primdahl et al (1999) have reported that β-catenin mutation is not found in 27 samples of microsatellite stable CRC. It can therefore be inferred that β-catenin mutation is implicated in only a very small proportion of CRC that may be a subset of HNPCC cases.
Recent experimental and clinical analyses have indicated that cytoplasmic expression and nuclear translocation of β-catenin in colorectal tumour cells usually results from mutations in the APC or β-catenin genes (Morin et al, 1997; Rubinfeld et al, 1997; Iwao et al, 1998). Unregulated intracellular β-catenin protein has been shown to activate its target gene expression by the complex formation with a family of nuclear transcription factors. APC mutations are present in about 80% of sporadic colorectal adenomas and cancers, while in recent studies (Uchida et al, 1998; Dehari, 2001), APC mutations have been rare in SAs. We observed no mutation of β-catenin in SAs. Nuclear or cytoplasmic expression of β-catenin was significantly less common in SAs than in CADs or SCAs. Furthermore, no nuclear or cytoplasmic immunostaining of β-catenin was observed in adjacent hyperplastic or conventionally adenomatous glands in mixed-type SAs. Thus, the signalling pathway involving APC and β-catenin seems unlikely to be important for tumorigenesis in most SAs.
The previous study (Samowitz et al, 1999) has suggested that β-catenin mutation occurs in a small subset of CAD, and CAD initiated by β-catenin mutation may rarely progress to CRC. From the preceding observation, it can be deduced that β-catenin is not implicated in the initiation of SAs, or that SAs rarely progress to carcinoma. We demonstrated that no mutation of β-catenin was detected in SAs. Furthermore, when we separately examined β-catenin mutation in serrated and nonserrated areas of mixed-type SAs, no β-catenin mutation was found in either area. Thus, mutations in exon 3 of β-catenin are unlikely to contribute to tumorigenesis in SA. In fact, it has been previously shown that β-catenin and APC mutations are uncommon in SAs (Sawyer et al, 2002). This supports the suggested role of SAs in the pathogenesis of sporadic MSI-H cancer.
We found one mixed-type SA with two histologically distinct areas representing carcinoma and SA. Although focally nuclear expression of β-catenin was demonstrated in the carcinomatous component of this lesion, cytoplasmic accumulation of β-catenin was observed. Neither nuclear nor cytoplasmic staining was found in the adjacent serrated epithelium. The immunohistologic heterogeneity demonstrated in this tumour implies that intracellular redistribution of β-catenin might be a late event in the development of colorectal cancer. However, only one such case of carcinoma associated with SA was analysed in this study, so further investigation of these genetic changes is required to clarify the mechanism of tumorigenesis in SA.
In summary, we evaluated the status of β-catenin in the development of SA using immunohistochemical and genetic methods. Intracellular localisation of β-catenin in SAs differed from that in CADs or SCAs, and no β-catenin mutation was observed in SAs. We concluded that β-catenin mutation is unlikely to contribute to tumorigenesis in SA. Further, intracellular localisation of β-catenin may not be associated with an early tumorigenic event in most SAs.
Change history
16 November 2011
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Ajioka Y, Watanabe H, Jass JR, Yokota Y, Kobayashi M, Nishikura K (1998) Infrequent K-ras codon 12 mutation in serrated adenomas of human colorectum. Gut 42: 680–684
Barth AI, Nathke IS, Nelson WJ (1997) Cadherins, catenins and APC protein: interplay between cytoskeletal complexes and signaling pathways. Curr Opin Cell Biol 9: 683–690
Behrens J, von Kries JP, Kuhl M, Bruhn L, Wedlich D, Grosschedl R, Birchmeier W (1996) Functional interaction of beta-catenin with the transcription factor LEF-1. Nature 382: 638–642
Brabletz T, Herrmann K, Jung A, Faller G, Kirchner T (2000) Expression of nuclear beta-catenin and c-myc is correlated with tumor size but not with proliferative activity of colorectal adenomas. Am J Pathol 156: 865–870
Dehari R (2001) Infrequent APC mutations in serrated adenoma. Tohoku J Exp Med 193: 181–186
Hao X, Tomlinson I, Ilyas M, Palazzo JP, Talbot IC (1997) Reciprocity between membranous and nuclear expression of beta-catenin in colorectal tumours. Virch Arch 431: 167–172
Hawkins NJ, Ward RL (2001) Sporadic colorectal cancers with microsatellite instability and their possible origin in hyperplastic polyps and serrated adenomas. J Natl Cancer Inst 93: 1307–1313
He TC, Sparks AB, Rago C, Hermeking H, Zawel L, da Costa LT, Morin PJ, Vogelstein B, Kinzler KW (1998) Identification of c-MYC as a target of the APC pathway. Science 281: 1509–1512
Hiyama T, Yokozaki H, Shimamoto F, Haruma K, Yasui W, Kajiyama G, Tahara E (1998) Frequent p53 gene mutations in serrated adenomas of the colorectum. J Pathol 186: 131–139
Hugh TJ, Dillon SA, Taylor BA, Pignatelli M, Poston GJ, Kinsella AR (1999) Cadherin-catenin expression in primary colorectal cancer: a survival analysis. Br J Cancer 80: 1046–1051
Iino H, Jass JR, Simms LA, Young J, Leggett B, Ajioka Y, Watanabe H (1999) DNA microsatellite instability in hyperplastic polyps, serrated adenomas, and mixed polyps: a mild mutator pathway for colorectal cancer? J Clin Pathol 52: 5–9
Inomata M, Ochiai A, Akimoto S, Kitano S, Hirohashi S (1996) Alteration of beta-catenin expression in colonic epithelial cells of familial adenomatous polyposis patients. Cancer Res 56: 2213–2217
Iwao K, Nakamori S, Kameyama M, Imaoka S, Kinoshita M, Fukui T, Ishiguro S, Nakamura Y, Miyoshi Y (1998) Activation of the beta-catenin gene by interstitial deletions involving exon 3 in primary colorectal carcinomas without adenomatous polyposis coli mutations. Cancer Res 58: 1021–1026
Jass JR, Burt R (2000) Hyperplastic polyposis. In: Hamilton SR, Aaltonnen LA, eds. WHO International Classification of Tumors (3rd edn): Pathology and Genetics of Tumors of the digestive system. Berlin: Springer-Verlag, 135–136
Jass JR, Iino H, Ruszkiewicz A, Painter D, Solomon MJ, Koorey DJ, Cohn D, Furlong KL, Walsh MD, Palazzo J, Edmonston TB, Fishel R, Young J, Leggett BA (2000a) Neoplastic progression occurs through mutator pathways in hyperplastic polyposis of the colorectum. Gut 47: 43–49
Jass JR, Young J, Leggett BA (2000b) Hyperplastic polyps and DNA microsatellite unstable cancers of the colorectum. Histopathology 37: 295–301
Kinzler KW, Vogelstein B (1996) Lessons from hereditary colorectal cancer. Cell 87: 159–170
Kitaeva MN, Grogan L, Williams JP, Dimond E, Nakahara K, Hausner P, DeNobile JW, Soballe PW, Kirsch IR (1997) Mutations in beta-catenin are uncommon in colorectal cancer occurring in occasional replication error-positive tumors. Cancer Res 57: 4478–4481
Kobayashi M, Honma T, Matsuda Y, Suzuki Y, Narisawa R, Ajioka Y, Asakura H (2000) Nuclear translocation of beta-catenin in colorectal cancer. Br J Cancer 82: 1689–1693
Konishi M, Kikuchi-Yanoshita R, Tanaka K, Muraoka M, Onda A, Okumura Y, Kishi N, Iwama T, Mori T, Koike M, Ushio K, Chiba M, Nomizu S, Konishi F, Utsunomiya J, Miyaki M (1996) Molecular nature of colon tumors in hereditary nonpolyposis colon cancer, familial polyposis, and sporadic colon cancer. Gastroenterology 111: 307–317
Korinek V, Barker N, Morin PJ, van Wichen D, de Weger R, Kinzler KW, Vogelstein B, Clevers H (1997) Constitutive transcriptional activation by a beta-catenin–Tcf complex in APC−/− colon carcinoma. Science 275: 1784–1787
Longacre TA, Fenoglio-Preiser CM (1990) Mixed hyperplastic adenomatous polyps/serrated adenomas. A distinct form of colorectal neoplasia. Am J Surg Pathol 14: 524–537
Makinen MJ, George SM, Jernvall P, Makela J, Vihko P, Karttunen TJ (2001) Colorectal carcinoma associated with serrated adenoma–prevalence, histological features, and prognosis. J Pathol 193: 286–294
Makino R, Kaneko K, Kurahashi T, Matsumura T, Mitamura K (2000) Detection of mutation of the p53 gene with high sensitivity by fluorescence-based PCR-SSCP analysis using low-pH buffer and an automated DNA sequencer in a large number of DNA samples. Mutat Res 452: 83–90
Matsumoto T, Mizuno M, Shimizu M, Manabe T, Iida M (1999) Clinicopathological features of serrated adenoma of the colorectum: comparison with traditional adenoma. J Clin Pathol 52: 513–516
Mirabelli-Primdahl L, Gryfe R, Kim H, Millar A, Luceri C, Dale D, Holowaty E, Bapat B, Gallinger S, Redston M (1999) Beta-catenin mutations are specific for colorectal carcinomas with microsatellite instability but occur in endometrial carcinomas irrespective of mutator pathway. Cancer Res 59: 3346–3351
Morin PJ, Sparks AB, Korinek V, Barker N, Clevers H, Vogelstein B, Kinzler KW (1997) Activation of beta-catenin–Tcf signaling in colon cancer by mutations in beta-catenin or APC. Science 275: 1787–1790
Otori K, Oda Y, Sugiyama K, Hasebe T, Mukai K, Fujii T, Tajiri H, Yoshida S, Fukushima S, Esumi H (1997) High frequency of K-ras mutations in human colorectal hyperplastic polyps. Gut 40: 660–663
Rashid A, Houlihan PS, Booker S, Petersen GM, Giardiello FM, Hamilton SR (2000) Phenotypic and molecular characteristics of hyperplastic polyposis. Gastroenterology 119: 323–332
Rubinfeld B, Albert I, Porfiri E, Munemitsu S, Polakis P (1997) Loss of beta-catenin regulation by the APC tumor suppressor protein correlates with loss of structure due to common somatic mutations of the gene. Cancer Res 57: 4624–4630
Salahshor S, Kressner U, Pahlman L, Glimelius B, Lindmark G, Lindblom A (1999) Colorectal cancer with and without microsatellite instability involves different genes. Genes Chromosomes Cancer 26: 247–252
Samowitz WS, Powers MD, Spirio LN, Nollet F, van Roy F, Slattery ML (1999) Beta-catenin mutations are more frequent in small colorectal adenomas than in larger adenomas and invasive carcinomas. Cancer Res 59: 1442–1444
Sawyer EJ, Cerar A, Hanby AM, Gorman P, Arends M, Talbot IC, Tomlinson IP (2002) Molecular characteristics of serrated adenomas of the colorectum. Gut 51: 200–206
Sparks AB, Morin PJ, Vogelstein B, Kinzler KW (1998) Mutational analysis of the APC/beta-catenin/Tcf pathway in colorectal cancer. Cancer Res 58: 1130–1134
Takayama T, Shiozaki H, Shibamoto S, Oka H, Kimura Y, Tamura S, Inoue M, Monden T, Ito F, Monden M (1996) Beta-catenin expression in human cancers. Am J Pathol 148: 39–46
Tetsu O, McCormick F (1999) Beta-catenin regulates expression of cyclin D1 in colon carcinoma cells. Nature 398: 422–426
Uchida H, Ando H, Maruyama K, Kobayashi H, Toda H, Ogawa H, Ozawa T, Matsuda Y, Sugimura H, Kanno T, Baba S (1998) Genetic alterations of mixed hyperplastic adenomatous polyps in the colon and rectum. Jpn J Cancer Res 89: 299–306
Valizadeh A, Karayiannakis AJ, el-Hariry I, Kmiot W, Pignatelli M (1997) Expression of E-cadherin-associated molecules (alpha-, beta-, and gamma-catenins and p120) in colorectal polyps. Am J Pathol 150: 1977–1984
Wong NA, Morris RG, McCondochie A, Bader S, Jodrell DI, Harrison DJ (2002) Cyclin D1 overexpression in colorectal carcinoma in vivo is dependent on beta-catenin protein dysregulation, but not K-ras mutation. J Pathol 197: 128–135
Acknowledgements
We thank Ms Hisako Nozawa and Ms Yoshiko Sasaki for excellent technical assistance and Dr Hideki Akita for helpful discussions.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Yamamoto, T., Konishi, K., Yamochi, T. et al. No major tumorigenic role for β-catenin in serrated as opposed to conventional colorectal adenomas. Br J Cancer 89, 152–157 (2003). https://doi.org/10.1038/sj.bjc.6601070
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.bjc.6601070
Keywords
This article is cited by
-
DNA methylation alterations of AXIN2 in serrated adenomas and colon carcinomas with microsatellite instability
BMC Cancer (2014)
-
Serrated polyps of the large intestine: current understanding of diagnosis, pathogenesis, and clinical management
Journal of Gastroenterology (2013)
-
BRAF mutations and phosphorylation status of mitogen-activated protein kinases in the development of flat and depressed-type colorectal neoplasias
British Journal of Cancer (2006)
-
Serrated adenoma of the colorectum and the DNA-methylator phenotype
Nature Clinical Practice Oncology (2005)
