
Abstract
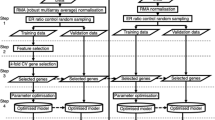
One common objective in microarray experiments is to identify a subset of genes that express differentially among different experimental conditions, for example, between drug treatment and no drug treatment. Often, the goal is to determine the underlying relationship between poor versus good gene signatures for identifying biological functions or predicting specific therapeutic outcomes. Because of the complexity in studying hundreds or thousands of genes in an experiment, selection of a subset of genes to enhance relationships among the underlying biological structures or to improve prediction accuracy of clinical outcomes has been an important issue in microarray data analysis. Selection of differentially expressed genes is a two-step process. The first step is to select an appropriate test statistic and compute the P-value. The genes are ranked according to their P-values as evidence of differential expression. The second step is to assign a significance level, that is, to determine a cutoff threshold from the P-values in accordance with the study objective. In this paper, we consider four commonly used statistics, t-, S- (SAM), U-(Mann–Whitney) and M-statistics to compute the P-values for gene ranking. We consider the family-wise error and false discovery rate false-positive error-controlled procedures to select a limited number of genes, and a receiver-operating characteristic (ROC) approach to select a larger number of genes for assigning the significance level. The ROC approach is particularly useful in genomic/genetic profiling studies. The well-known colon cancer data containing 22 normal and 40 tumor tissues are used to illustrate different gene ranking and significance level assignment methods for applications to genomic/genetic profiling studies. The P-values computed from the t-, U- and M-statistics are very similar. We discuss the common practice that uses the P-value, false-positive error probability, as the primary criterion, and then uses the fold-change as a surrogate measure of biological significance for gene selection. The P-value and the fold-change can be pictorially shown simultaneously in a volcano plot. We also address several issues on gene selection.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 6 print issues and online access
$259.00 per year
only $43.17 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout



Similar content being viewed by others


References
Simon R, Wang SJ . Use of genomic signatures in therapeutics development in oncology and other diseases. The Pharmacogenom J 2006; 6: 166–173.
Smyth GK, Yang YH, Speed TP . Statistical issues in cDNA microarray data analysis. In: Brownstein MJ, Khodursky A (eds) Functional Genomics: Methods and Protocols. Methods in Molecular Biology. Humana Press: Totowa, NJ, 2003; 224: 111–136.
Golub T, Slonim D, Tamayo P, Huard C, Gassenbeek M, Mesirov J et al. Molecular classification of cancer: class discovery and class prediction by gene expression monitoring. Science 1999; 286: 531–537.
Tsai CA, Chen CH, Lee TC, Ho IC, Yang UC, Chen JJ . Gene Selection for sample classifications in microarray experiments. DNA Cell Biol 2004; 23: 607–614.
Tusher VG, Tibshirani R, Chu G . Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci USA 2001; 98: 5116–5121.
Efron B, Tibshirani R, Storey J, Tusher V . Empirical bayes analysis of a microarray experiment. J Am Stat Assoc 2001; 96: 1151–1160.
Tsai CA, Chen YJ, Chen JJ . Testing for differentially expressed genes with microarray data. Nucleic Acids Res 2003; 31: e52.
Westfall PH, Young SS . Resampling-Based Multiple Testing. John Wiley & Sons: New York, 1993.
Benjamini Y, Hochberg Y . Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc B 1995; 57: 289–300.
Hsueh H, Chen JJ, Kodell RL . Comparison of methods for estimating number of true null hypothesis in multiplicity testing. J Biopharm Stat 2003; 13: 675–689.
Hsueh H, Tsai CA, Chen JJ . Incorporating the number of the true hypotheses to improve power in multiple testing: application to gene microarray data. J Stat Comput Simulation, to appear.
Delongchamp RR, Bowyer JF, Chen JJ, Kodell RL . Multiple testing strategy for analyzing cDNA array data on gene expression. Biometrics 2004; 60: 774–782.
Alon U, Barkai N, Notterman DA, Gish K, Ybarra S, Mack D et al. Broad patterns of gene expression revealed by clustering analysis of tumor and normal colon tissues probed by oligonucleotide arrays. Proc Natl Acad Sci USA 1999; 96: 6745–6750.
Jin W, Riley RM, Wolfinger RD, White KP, Passador-Gurgel G, Gibson G . The contributions of sex, genotype and age to transcriptional variance in Drosophila melanogaster. Nat Genet 2001; 29: 389–395.
Qin LX, Kerr KF . Contributing Members of the Toxicogenomics Research Consortium. Empirical evaluation of data transformations and ranking statistics for microarray analysis. Nucleic Acids Res 2004; 32: 5471–5479.
van’t veer LJ, Dai H, van de vijver MJ, He YD, Hart AAM, Mao M et al. Gene expression profiling predicts clinical outcome of breast cancer. Nature 2002; 415: 530–536.
Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, Lehar J et al. PGC-1α-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet 2003; 34: 267–273.
Larkin JE, Frank BC, Gavas H, Sultana R, Quackenbush J . Independence and reproducibility across microarray platforms. Nat Methods 2005; 2: 337–343.
Irizarry RA, Warren D, Spencer F, Kim IF, Biswal S, Frank BC et al. Multiple-laboratory comparison of microarray platforms. Nat Methods 2005; 2: 345–349.
Members of the Toxicogenomics Research Consortium. Standardizing global gene expression analysis between laboratories and across platforms. Nat Methods 2005; 2: 351–356.
Tsai CA, Wang SJ, Chen DT, Chen JJ . Sample size for gene expression microarray experiments. Bioinformatics 2005; 21: 1502–1508.
Author information
Authors and Affiliations
Corresponding author
Additional information
The views presented in this paper are those of the authors and do not necessarily represent those of the US Food and Drug Administration.
Rights and permissions
About this article
Cite this article
Chen, J., Wang, SJ., Tsai, CA. et al. Selection of differentially expressed genes in microarray data analysis. Pharmacogenomics J 7, 212–220 (2007). https://doi.org/10.1038/sj.tpj.6500412
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.tpj.6500412
Keywords
This article is cited by
-
Infants’ cortex undergoes microstructural growth coupled with myelination during development
Communications Biology (2021)
-
IL-36γ (IL-1F9) Is a Biomarker for Psoriasis Skin Lesions
Journal of Investigative Dermatology (2015)
-
Reactive oxygen species and transcript analysis upon excess light treatment in wild-type Arabidopsis thaliana vs a photosensitive mutant lacking zeaxanthin and lutein
BMC Plant Biology (2011)
-
Expression of the H+-ATPase AHA10 proton pump is associated with citric acid accumulation in lemon juice sac cells
Functional & Integrative Genomics (2011)
-
Functional modules with disease discrimination abilities for various cancers
Science China Life Sciences (2011)
