
Abstract
Several mutations in the melanocortin receptor 4 gene have been identified in humans and account for 3–6% of morbid obesity. In contrast, strong evidence of a causative role for melanocortin receptor 3 (MC3R) mutations are still lacking. In MC3R knockout mice, high feed efficiency rather than hyperphagia seems to contribute to increased fat mass. On the basis of this evidence, the objective of the present study was to investigate the presence of MC3R mutations in a group of 290 obese subjects (mean BMI 44.2±5.9 kg/m2). As a control, a group of 215 normal-weight subjects (mean BMI 22.4±2.7 kg/m2) was also screened. Three novel mutations in the MC3R gene (A293T, I335S and X361S) were identified among the obese patients. The mutations segregated with obesity in the members of the families studied. In vitro expression studies of each mutation demonstrated a loss of function of the I335S-mutated receptor. These findings suggest that, in humans, MC3R mutations may be a cause of a dominantly inherited form of obesity. However, this association as well as the specific phenotypic characteristics resulting from these mutations need to be further evaluated in larger series of obese subjects.
Similar content being viewed by others

Introduction
Obesity is a multifactorial disease with a strong genetic component.1 The recent advances in rodent genetics2 have led to the identification of a number of molecules participating in the pathways that regulate energy homeostasis. Mutations in the genes coding for these molecules are responsible for some monogenic forms of obesity both in mice3, 4, 5, 6, 7 and humans.8, 9, 10, 11, 12, 13, 14 Among these genes, melanocortin receptors 3 and 4 (MC3R and MC4R) have been studied for their localization in the hypothalamus and their role in the leptin signalling pathway.14 In mice, inactivation of either receptor leads to obesity with the double knockout phenotype suggesting that MC3R and MC4R have a nonredundant role.6 Several mutations of the MC4R gene have been described in humans and are associated, especially in children, with a specific obese phenotype characterized by early-onset obesity, hyperphagic behaviour, normal metabolic rate and increased fat and lean mass.15 The knockout mouse for MC3R is hypophagic, has a normal metabolic rate but shows a peculiar phenotype of decreased linear growth, increased fat mass and reduced lean mass. Thus, high feed efficiency, defined as the ratio of weight gain to food intake (FI), and not hyperphagia seems to contribute to the increased fat mass.6 Despite the detailed characterization of the obese phenotype in the MC3R knockout mice, strong evidence that mutations in the homologous human gene may be a cause of obesity are still lacking. Indeed, to date only two variants16 and one mutation17 have been described in the MC3R-coding region. In the populations examined, the presence of the T6K (C17A) and the V81I (G241A) variants was not associated with any phenotypic characteristic of obesity. Only recently, a study performed on 355 overweight and nonoverweight children indicated that 8.2% were double homozygous for the two variants and had significantly higher BMI and body fat compared with wild-type (WT) and heterozygous children.18 In contrast, the I183N (T548A) mutation, which was not found in control subjects17 and was functionally inactive in vitro,19, 20 did not segregate with obesity in the family studied.17
Thus, to further investigate the possible role of MC3R alterations in the pathogenesis of obesity, in the present study we screened the entire coding region of the MC3R gene in 290 severely obese subjects.
Materials and methods
Subjects
The present study was conducted on 290 obese patients referred to the Division of General Medicine of the San Giuseppe Hospital, Istituto Auxologico Italiano (Piancavallo, Italy), for diagnostic or therapeutic problems related to obesity or its morbidity. The patient group included 205 women and 85 men, 40.0±11.9 years (mean±SD), with a mean BMI of 44.2±5.9 kg/m2. Anthropometric variables measured included body weight, height, waist and hip circumference. Body composition, in terms of the percentage of fat body mass and fat-free body mass, was determined by bioelectrical impedance analysis (BIA101/S model; Akern, Florence, Italy). Patients with fluid overload, according to vectorial analysis, were excluded to minimize errors in estimating fat body mass and fat-free body mass in severe obesity.21 Average daily caloric intake was evaluated by a 7-day recall standardized technique, details of which have been published previously.22 The interviews were always carried out by two different trained dieticians, who used a food frequency questionnaire and an atlas for assessment of food-portion size. Moreover, all obese patients showing a negative difference between reported food intake and resting energy expenditure (REE) multiplied by 1.2 (as a cautious estimate of total energy expenditure) were considered as underreporting and, thus, their case history was classified as unreliable.22 Diet and weight histories were assessed by interview according to standardized methodologies.23 REE (kcal/24 h) was determined following an overnight fast and at rest in a thermoregulated room (22–24°C) using computed open-circuit indirect calorimetry (Sensormedics, Milan, Italy). Resting oxygen uptake and resting carbon dioxide production were measured by a ventilated canopy at 1-min intervals for 30 min and expressed as a 24-h value. Blood samples were obtained for genomic DNA extraction from the obese subjects and 215 normal-weight control subjects 36.0±12 years old with mean BMI of 22.4±2.7 kg/m2. The study protocol was approved by the Institution Ethics Committee. The aim and the design of the study were explained to the patients who gave their informed consent.
Direct nucleotide sequencing of MC3R gene
Two set of primers (MC3R-A, 5′-GGGAGACAGAAGGAAGACAGC-3′ and MC3R-R1, 5′-GACGCCGCAGCAGACCCAGAT-3′; MC3R-C, 5′-TGCAACCTCCTGGCCATCGC-3′ and MC3R-R2, 5′-CGTGGATGGAAAGTCAAAAGT-3′) were used in a PCR to amplify two overlapping fragments covering the entire MC3R-coding region. The PCR was performed using the following conditions: denaturation at 95°C for 5 min, followed by 35 cycles of denaturation at 95°C for 45 s, annealing at 55°C for 45 s and elongation at 72°C for 1 min followed by a final elongation of 7 min. The PCR fragments were sequenced using the BigDye Teminator Kit (Applied Biosystem, Foster City, CA, USA) with primers MC3R-B (5′-TCTCTCTACCCTCCCCATCC-3′), MC3R-R1, MC3R-C and MC3R-R2 and analysed on the ABI Prism 310 automated sequencer (Applied Biosystem).
The normal-weight control population was genotyped for the three mutations by allele-specific PCR or restriction fragment length polymorphism analysis. The presence of A293T and I335S mutations was evaluated by PCR with MC3R-C and MC3R-R2 primers followed by an ApaI or DdeI digestion. The A293T mutation causes the loss of the ApaI restriction site, while I335S mutation creates a new DdeI restriction site. To evaluate the presence of X361S mutation, we performed an allele-specific PCR with a common primer MC3R-C and a specific primer MC3R-stop (5′-TTTCCATGGCCCTGCATCCT-3′) or MC3R-Ser (5′-TTTCCATGGCCCTGCATCCG-3′).
Cloning of wild-type and mutated MC3R
The entire coding region of MC3R gene was amplified using MC3R-B and MC3R-R2 primers and the PCR product was cloned into a pTarget Mammalian Expression vector (Promega, Madison, WI, USA). The three mutated constructs were obtained by site-directed mutagenesis of MC3R WT construct using the QuickChange Site-Directed Mutagenesis Kit (Stratagene, La Jolla, CA, USA). The WT sequence and the presence of mutations were confirmed by sequence analysis.
Intracellular cAMP determination
Ligand-stimulated receptor activity was evaluated by determining the intracellular cyclic 3′,5′-adenosine monophosphate (cAMP) content after stimulation with α-melanocortin stimulating hormone (α-MSH). COS7 cells were maintained in DMEM supplemented with 10% calf serum (HyClone, Logan, UT, USA), L-glutamine and penicillin/streptomycin. The day before transfection, 300 000 cells/well were plated in six-well plates. The following day the cells were transfected with FuGene Trasfection Reagent (Roche Diagnostics, Basel, Switzerland) according to the manufacturer's protocol. After 48 h, cells were washed and incubated in a stimulation medium composed of DMEM with 0.1 mg/ml BSA and 0.25 mM IBMX and varying concentrations of α-MSH (Sigma, St Louis, MO, USA) for 1 h at 37°C in a 5% CO2 incubator. Following the incubation, the media was replaced with 1 N HCl. An aliquot of the cell lysate was used to determine cAMP content using a commercially available radioimmunoassay (Perkin Elmer, Wellesley, MA, USA). Each experiment was performed in triplicate and repeated at least three times.
Cellular localization of WT and mutated MC3Rs
The WT and mutant MC3Rs were subcloned in GFP N-term Fusion TOPO TA vector (Invitrogen, Carlsbad, CA, USA). COS7 cells were transfected with either the WT or mutant MC3Rs as described previously. Forty-eight hours after transient transfection, the cells were washed with PBS (Invitrogen), fixed with 2% paraformaldehyde and mounted with Vectashield mounting medium (Vector Laboratories, Peterborough, England). The cells were visualized via confocal microscope (Bio-Rad, Hercules, CA, USA).
Results
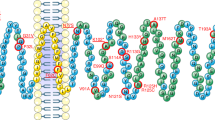
In all the 290 obese patients, the entire coding region of the MC3R gene was screened by automatic direct sequencing. We identified subjects carrying the already reported T6K and the V81I variants, which showed a near complete linkage disequilibrium. Their allelic frequencies were 8.6 and 7.6% in line with those detected in other Caucasian cohorts studied.18, 24, 25, 26 Interestingly, we detected three subjects heterozygous for three novel mutations that were not present in the 215 normal-weight control subjects. The possible presence of concomitant MC4R mutations was excluded by direct sequencing of the coding region of the MC4R gene (data not shown). Patient A was a man of 67 years with a BMI of 46. He carried an A293T (G877A) change in the sixth transmembrane domain of the protein. He was affected by NIDDM and osteoporosis, and died during the course of the study. Patient B was a woman of 56 years with a BMI of 39. In her clinical history, she had only a hypothyroidism due to autoimmune thyroiditis, which was successfully treated with replacement therapy. It must be underlined that her weight gain preceded the development of the hypothyroidism and was not affected by the replacement therapy. She carried a I335S (T1004G) amino-acid change in the final transmembrane domain of the receptor. Patient C was a woman of 22 years with a BMI of 44.3, carrying a X361S (A1082C) mutation that abolished the stop codon and led to the addition of seven extra amino acids to the intracellular C terminus of the receptor (SDAGPWL). Table 1 compares some clinical and anthropometric parameters of the three carriers with those of the entire obese population screened. It is noteworthy that the patients carrying MC3R mutations had markedly lower FI compared with that of the group of the other obese subjects studied while their REE was not reduced. Their REE/FI ratio was higher than 0.7, suggesting that they are able to use less than 30% of their FI for living activities. The REE/FI ratio was then evaluated in all the obese subjects studied, and this analysis revealed that among them there were additional 72 patients showing an REE/FI ratio between 0.7 and 0.83. Thus, there seems to exist a subgroup of obese patients in which obesity may be not associated with an excessive caloric intake.
We subsequently examined the co-segregation of these mutations in the families of patients B and C. In the first family, we were able to genotype the brother and sister of the proband (Figure 1). The brother was 57 years old with a BMI of 30, a history of early-onset obesity and strong resistance to lose weight, despite continuous dieting. He was heterozygous for the same mutation carried by the proband. In contrast, the 62-year-old sister had a BMI of 26, no history of obesity and was WT. In the second family, both parents were investigated: the obese father (BMI 38) was heterozygous for the X361S mutation, while the overweight mother was WT (Figure 1).

Pedigrees and genotypes of the family members of patients B and C. Filled symbols indicate affected individuals (BMI>30 kg/m2) with arrows indicating patients B and C.
To evaluate the functional activity of the mutated receptors, in vitro expression studies were performed. The WT and mutated receptors were transiently transfected in COS7 cells and the intracellular concentrations of cAMP, following stimulation with increasing doses of α-MSH, were measured (Figure 2). The A293T and X361S mutations did not impair the cAMP response with the EC50 (2.14 and 1.25 nM), similar to that obtained for the WT-transfected cells (1.82 nM). In contrast, the I335S mutation led to a complete loss of function of the receptor. We then investigated the subcellular localization of the WT and mutated receptors (Figure 3). The A293T and X361S mutations did not alter the normal cell surface expression like WT-MC3R, whereas COS7 cells transfected with the I335S-mutated receptor showed a diffuse cytoplasmic staining indicating an intracellular retention of the protein.

Functional studies of MC3R mutations. COS7 cells were transiently transfected for 48 h with WT and mutated receptors cloned into the pTarget expression vector, and subsequently stimulated with increasing concentrations of α-MSH (0–10−7 M) in serum-free conditions. Intracellular cAMP levels were measured by RIA with results showing the mean±SEM cAMP (pmol/well) production. Transfections were performed in triplicate and the experiments were repeated at least three time with similar results.

Subcellular localization studies of MC3R mutants. COS7 cells were transiently transfected with WT and mutated receptors cloned into the GFP Fusion TOPO TA vector for 42 h and analysed by confocal microscopy. Nuclear staining was performed with propidium iodide (1 μg/ml). White arrows indicate the presence of fluorescence on the cytoplasmic membranes.
Discussion
To the best of our knowledge, this is the first study that reports, in adult obese subjects, genetic and functional evidence for an association of MC3R mutations with morbid obesity. Moreover, as already reported for MC4R,27 these findings also indicate that in humans MC3R may represent a second locus at which mutations cause dominantly inherited forms of obesity.
The central role of melanocortins as catabolic mediators of leptin actions in the regulation of energy balance has been well established.14 It is now known that in the hypothalamus, α-MSH is produced by cleavage of pro-opiomelanocortin peptide and acts on the two neuronal isoforms of the melanocortin receptors: MC3R and MC4R. The relative importance of these isoforms in mediating the catabolic effects of melanocortins has not been completely elucidated. Indeed, MC4R seems to be critical for normal control of FI as inactivating mutations of its gene, both in mice and humans, led to an obese phenotype characterized by hyperphagia.5, 15 In contrast, MC3R seems to be implicate in the control of fat storage as MC3R−/− and MC3R−/+ mice are hypophagic and have increased feed efficiency when compared with WT littermates.6 Despite these detailed animal studies, to date the role of MC3R in humans has not been clearly defined. Several studies in different cohorts of obese subjects have identified two common variants in the coding region of the gene16, 24 and the homozygosity for these two variants has recently been associated to an increased BMI, although no data on feed efficiency were reported in this study.18 We also have identified the T6K and the V81I variants in our population with allelic frequencies in line with those reported in other studies in Caucasian subjects.18, 24, 25, 26 The single mutation so far identified for MC3R does not segregate with obesity.17 Thus, the results of the present study contribute to gaining insights into the functional role of MC3R in regulating energy homeostasis in humans. It is noteworthy that the three carrier patients belong to a subgroup of obese subjects reporting a low FI in relation to their REE. This characteristic is highlighted by the REE/FI ratio, whose values comprised between 0.7 and 0.83. We must underline that data on FI were reported by the patients and thus, although great care was used in performing the interviews and in excluding patients with an REE/FI>0.83, underreporting cannot be completely ruled out. Taken together, the data in the animal model and the results of the present study, it is tempting to speculate that there could exist a new MC3R-related monogenic form of severe obesity, which is not primarily caused by an increased FI. This hypothesis needs to be further investigated in larger series of obese subjects in which caloric intake is more precisely documented.
Although we could investigate only a limited number of family members, these studies suggest a co-segregation of the identified mutations with the obese phenotype. Furthermore, the I335S mutant resulted in a complete loss of function of the receptor most likely due to the absence of cell surface expression. Interestingly, based on the alignment of the amino-acid sequences of the melanocortin receptor family members, reported in the GPCR's Database (http://www.gpcr.org/7tm/), we observed that the I335S mutations are localized in a highly conserved region of the receptor and that the aa I335 in MC3R corresponds to aa I301 of MC4R. An I301T substitution in MC4R has been described by Vaisse et al27 as a loss-of-function mutation, thus suggesting a critical role of this amino-acid position for the full biological function of both melanocortin receptors. This is also confirmed by the results of a bioinformatic approach. Using SIFT, PANTHER and POLYPHEN, a computational prediction of the functional effects of an amino-acid exchange, as was the I335S and the A293T substitution, resulted in a nonfunctioning protein.
There are alternate hypotheses to explain why the A293T and X361S mutants did not show an altered functional activity in the present studies. It is well-known that MC3R can bind to both γ-MSH as well as α-MSH,28 thus these in vitro conditions may not reflect the real biological properties of the receptor. Moreover, it has been previously reported that MC3R is involved in other intracellular signalling pathways including protein kinase C and inositol three phosphate.29, 30 However, until these additional mechanisms will be investigated, we cannot definitively exclude that these genetic variants are only polymorphisms, and, thus, further studies are needed before any conclusion can be drawn regarding their role in the development of obesity.
In conclusion, taken together with the study in the animal model, the results presented herein support the hypothesis that in humans also mutated MC3R might play a causative role for dominantly inherited monogenic forms of obesity. To confirm the linkage of these mutations with obesity in larger pedigrees and to obtain a detailed phenotypic characterization of these forms, we plan to further extend these studies in larger series of morbid obese Italian patients.
References
O’Rahilly S, Farooqi IS, Yeo GSH, Challis BG : Minireview: human obesity – lessons from monogenic disorders. Endocrinology 2003; 144: 3757–3764.
Leibel RL, Chung WK, Chua Jr SC : The molecular genetics of rodent single gene obesities. J Biol Chem 1997; 272: 31937–31940.
Miller MW, Duhl DM, Vrieling H et al: Cloning of the mouse agouti gene predicts a secreted protein ubiquitously expressed in mice carrying the lethal yellow mutation. Genes Dev 1993; 7: 454–467.
Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman JM : Positional cloning of the mouse obese gene and its human homologue. Nature 1994; 372: 425–432.
Huszar D, Lynch CA, Fairchild-Huntress V et al: Targeted disruption of the melanocortin-4 receptor results in obesity in mice. Cell 1997; 88: 131–141.
Chen AS, Marsh DJ, Trumbauer ME et al: Inactivation of the mouse melanocortin-3 receptor results in increased fat mass and reduced lean body mass. Nat Genet 2000; 26: 97–102.
Zhu X, Zhou A, Dey A et al: Disruption of PC1/3 expression in mice causes dwarfism and multiple neuroendocrine peptide processing defects. Proc Natl Acad Sci USA 2002; 99: 10293–10298.
Jackson RS, Creemers JW, Ohagi S et al: Obesity and impaired prohormone processing associated with mutations in the human prohormone convertase 1 gene. Nat Genet 1997; 16: 303–306.
Montague CT, Farooqi IS, Whitehead JP et al: Congenital leptin deficiency is associated with severe early-onset obesity in humans. Nature 1997; 387: 903–908.
Clément K, Vaisse C, Lahlou N et al: A mutation in the human leptin receptor gene causes obesity and pituitary dysfunction. Nature 1998; 392: 398–401.
Krude H, Biebermann H, Luck W, Horn R, Brabant G, Gruters A : Severe early-onset obesity, adrenal insufficiency and red hair pigmentation caused by POMC mutations in humans. Nat Genet 1998; 19: 155–157.
Vaisse C, Clément K, Guy-Grand B, Froguel P : A frameshift mutation in human MC4R is associated with a dominant form of obesity. Nat Genet 1998; 20: 113–114.
Yeo GS, Farooqi IS, Aminian S, Halsall DJ, Stanhope RG, O’Rahilly S : A frameshift mutation in MC4R associated with dominantly inherited human obesity. Nat Genet 1998; 20: 111–112.
Cummings DE, Schwartz MW : Genetics and pathophysiology of human obesity. Annu Rev Med 2003; 54: 453–471.
Farooqi IS, Keogh JM, Yeo GS, Lank EJ, Cheetham T, O’Rahilly S : Clinical spectrum of obesity and mutations in the melanocortin 4 receptor gene. N Engl J Med 2003; 348: 1085–1095.
Li WD, Joo EJ, Furlong EB et al: Melanocortin 3 receptor (MC3R) gene variants in extremely obese women. Int J Obesity 2000; 24: 206–210.
Lee YS, Kok-Seng L, Poh Kah-Yin L : A novel melanocortin 3 receptor gene (MC3R) mutation associated with severe obesity. J Clin Endocrinol Metab 2002; 87: 1423–1426.
Feng N, Young SF, Aguilera G et al: Co-occurrence of two partially inactivating polymorphisms of MC3R is associated with pediatric-onset obesity. Diabetes 2005; 54: 2663–2667.
Rached M, Buronfosse A, Begeot M, Penhoat A : Inactivation and intracellular retention of the human I183N mutated melanocortin 3 receptor associated with obesity. Biochem Biophys Acta 2004; 1689: 2229–2234.
Tao YX, Segaloff D : Functional characterization of melanocortin-3 receptor variants identify a loss-of-function mutation involving an amino acid critical for G protein-coupled receptor activation. J Clin Endocrinol Metab 2004; 89: 3936–3942.
Piccoli A, Brunani A, Savia G et al: Discriminating between body fat and fluid changes in the obese adult using bioimpedance vector analysis. Int J Obes Relat Metab Disord 1998; 22: 97–104.
Liuzzi A, Savia G, Tagliaferri M et al: Serum leptin concentration in moderate and severe obesity: relationship with clinical, anthropometric and metabolic factors. Int J Obesity 1999; 23: 1066–1073.
Wadden TA, Bartket S, Letizia KA, Foster GD, Stunkard AJ : Relationship of dieting history to resting metabolic rate, body composition, eating behaviour, and subsequent weight loss. Am J Clin Nutr 1992; 56: 203S–208S.
Hani EH, Dupont S, Durand E et al: Naturally occurring mutations in the melanocortin receptor 3 gene are not associated with type 2 diabetes mellitus in French Caucasians. J Clin Endocrinol Metab 2001; 86: 2895–2898.
Schalin-Jantti C, Valli-Jaakola K, Oksanen L et al: Melanocortin-3-receptor gene variants in morbid obesity. Int J Obes Relat Metab Disord 2003; 27: 70–74.
Boucher N, Lanouette CM, Larose M, Perusse L, Bouchard C, Chagnon YC : A +2138InsCAGACC polymorphism of the melanocortin receptor 3 gene is associated in human with fat level and partitioning in interaction with body corpulence. Mol Med 2002; 8: 158–165.
Vaisse C, Clément K, Durand E, Hercberg S, Guy-Grand B, Froguel P : Melanocortin-4 receptor mutations are a frequent and heterogeneous cause of morbid obesity. J Clin Invest 2000; 106: 253–262.
Gantz I, Konda Y, Tashiro T et al: Molecular cloning of a novel melanocortin receptor. J Biol Chem 1993; 268: 8246–8250.
Konda Y, Gantz I, DelValle J, Shimoto Y, Miwa H, Yamada T : Interaction of dual intracellular signaling pathways activated by the melanocortin-3 receptor. J Biol Chem 1994; 269: 13162–13166.
Wachira SJ, Hughes-Darden CA, Taylor CV, Ochillo R, Robinson TJ : Evidence for the interaction of protein kinase C and melanocortin 3-receptor signaling pathways. Neuropeptides 2003; 37: 201–210.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Mencarelli, M., Walker, G., Maestrini, S. et al. Sporadic mutations in melanocortin receptor 3 in morbid obese individuals. Eur J Hum Genet 16, 581–586 (2008). https://doi.org/10.1038/sj.ejhg.5202005
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.ejhg.5202005
Keywords
This article is cited by
-
Evaluation of the MC3R gene pertaining to body weight and height regulation and puberty development
Scientific Reports (2023)
-
Pharmacological effect of human melanocortin-2 receptor accessory protein 2 variants on hypothalamic melanocortin receptors
Endocrine (2018)
-
Effects of Melanocortin 3 and 4 Receptor Deficiency on Energy Homeostasis in Rats
Scientific Reports (2016)
-
Evidence for natural selection at the melanocortin-3 receptor gene in European and African populations
Acta Diabetologica (2016)
-
Prevalence of rare MC3R variants in obese cases and lean controls
Endocrine (2013)
