
Abstract
Variable transcription of the cardiac sodium channel gene is a candidate mechanism determining arrhythmia susceptibility. We have previously cloned and characterized the core promoter and flanking region of SCN5A, encoding the cardiac sodium channel. Loss-of-function mutations in this gene have been reported in ∼20% of patients with Brugada syndrome, an inherited cardiac electrical disorder associated with a high incidence of life-threatening arrhythmias. In this study, we identified DNA variants in the proximal 2.8 kb promoter region of SCN5A and determined their frequency in 1121 subjects. This population consisted of 88 Brugada syndrome patients with no SCN5A coding region mutation, and 1033 anonymized subjects from various ethnicities. Variant promoter activity was assayed in CHO cells and neonatal cardiomyocytes by transient transfection of promoter–reporter constructs. Single-nucleotide polymorphisms (SNPs) were identified at ∼1/200 base pairs which are: 11 in the 5′-flanking region, 1 in exon 1, and 5 in intron 1. In addition, a haplotype consisting of two SNPs in complete linkage disequilibrium was identified. Minor allele frequencies were >5% in at least one ethnic panel at 5/19 polymorphic sites. In vitro functional analysis in cardiomyocytes identified four variants with significantly (P<0.05) reduced reporter activity (up to 63% reduction). The largest changes were seen with c.−225−1790 G>A, which reduced reporter activity by 62.8% in CHO cells and 55% in cardiomyocytes. From these results, we can conclude that the SCN5A core promoter includes multiple DNA polymorphisms with altered in vitro activity, further supporting the concept of interindividual variability in transcription of this cardiac ion channel gene.
Similar content being viewed by others

Introduction
The SCN5A gene encodes the α-subunit of the predominant voltage-sensitive sodium channel expressed in cardiac muscle. The channel is an integral membrane protein that selectively allows sodium influx into the heart cell, propagates the initial upstroke of the action potential, and underlies fast conduction in human atrium and ventricle.1, 2 The human gene has been mapped to chromosome 3p21,3 and ‘gain of function’ mutations associated with defective fast inactivation cause type 3 of the congenital long QT syndrome.4 Loss-of-function mutations have also been described and linked to congenital arrhythmia syndromes, including conduction system failure,5 sick sinus syndrome,6 atrial fibrillation,7 and Brugada syndrome (BS),8 which predisposes to ventricular fibrillation (VF); in BS, SCN5A-coding region variants can be identified in ∼20–30% of affected patients. Loss of sodium channel function has been associated with an increased risk for VF not only in BS, but also with acquired lesions, such as acute myocardial ischemia9, 10 or therapy with sodium channel blocking drugs.9, 10, 11 Experimental evidence supports the idea that loss of sodium channel function can cause re-entrant excitation by slowing conduction12 or by enhancing heterogeneity of repolarization.13, 14, 15 Taken together, therefore, these data suggest that loss of sodium channel function is a common mechanism predisposing to life-threatening cardiac arrhythmias. For that reason, variable SCN5A transcription could be a candidate modulator of arrhythmia risk, particularly in the presence of added factors such as myocardial ischemia, drugs, or mutations. However, the role of DNA variants within SCN5A transcriptional control has not been yet thoroughly investigated.16, 17
Cis-acting elements are usually conserved among species and functionally organized into modules in which each module integrates input from a specific set of transcription factors to direct a corresponding spatio–temporal expression pattern.18, 19 While many regions of DNA may include regulatory functions, promoter elements are sites of assembly for transcription factors through a site-selection process that plays a central role in transcriptional regulation.20, 21 One recent promoter screening and functional study indicated that around a third of promoter variants may alter gene expression to a functionally relevant extent.22 We have previously cloned and characterized the core promoter and flanking region of human and mouse SCN5A genes, and reported a single-nucleotide polymorphism (SNP), c.−225−92 C>A (numbering see Table 2), present in 6/142 normal alleles, which increased transcriptional activity by ∼50% in cardiomyocytes.23 In addition, we have reported a six-change haplotype variant within this region that is common in east Asian populations, which reduces transcriptional activity, and is associated with conduction slowing in human populations.24 To further evaluate the functional effect of DNA variants in this region, we screened it for DNA variants in 88 patients with BS but no SCN5A-coding mutations, and 1033 control subjects of diverse ethnicities. We report here the frequency of these variants and their functional characterization, compared to wild-type, in promoter–reporter experiments.
Methods
Study populations
Patients
The BS was diagnosed in 88 patients following standard published criteria.25, 26 Cases are from multiple medical centers that include the Vanderbilt University Medical Center (n=13), the Academic Medical Center, University of Amsterdam (n=38), the University Hospital Münster (n=27), and the LMU University Clinics Großhadern, Munich (n=10). A blood sample was obtained from each patient for extraction of DNA from lymphocytes. DNA was then harvested and archived. The investigation conforms with the principles outlined in the Declaration of Helsinki and informed consent was obtained from all individuals. All patients had been previously screened for SCN5A-coding region mutations and no mutation had been identified.
Reference populations
Four reference populations were studied:
-
1
A set of randomly selected and unrelated Dutch Caucasian individuals (n=98).
-
2
Randomly selected and unrelated individuals from the general population living in Southern Germany, surveyed during 2002 and 2003 (n=702, from the KORA S4 survey, a population-based sub-survey originating from the WHO MONICA project).27
-
3
A set of randomly selected, unrelated individuals with ethnicities representing those of the middle Tennessee area (n=71; 48 white and 23 black).
-
4
A subset of ethnicity-defined but otherwise anonymized individuals (n=162, Coriell Polymorphism Discovery Resource multi-ethnicity panels that is available from the National Human Genome Research Institute).28 The Coriell samples used in this study included four different ethnicity panels, consisting of unrelated and apparently healthy individuals. The panels screened in this study were as follows: (a) the Caucasian Panel (self-declared Caucasians), n=47; (b) the African American Panel (individuals of self-declared African ancestry or 3/4 grandparents with African ancestry), n=45; (c) Han Chinese from Los Angeles Panel (individuals with all four grandparents born in Taiwan, China, or Hong Kong), n=26; and (d) the Mexican–American Community of Los Angeles Panel (individuals with three or four grandparents born in Mexico), n=44.
Thus, a total of 2066 control alleles and 176 alleles in patients with BS were studied.
Screening for variants
Polymorphism identification
Polymorphism identification was carried out in the BS patient group (n=88) and the Coriell group (n=162). The screened segment included 2.1 kb of 5′ upstream sequence of exon 1, exon 1 (which is 173 bp and non-coding), and the proximal intron 1 regions that are relatively GC-rich (60.6% GC content) and highly conserved compared to mouse and rat. Proximal intron 1 of 439 bp was screened in the patient group, while in the Coriell group, the screening was extended to 708 bp of proximal intron 1. A 2.8 kb PCR fragment encompassing these regions was amplified with primer pair 1F/1R (Table 1), using the LA-PCR method (LA PCR kit, TaKaRa). Thermal cycling conditions were 94°C for 1 min, followed by 40 cycles of 94°C for 30 s, 64°C for 30 s, and 68°C for 3 min. The intron 1 fragment was amplified with primer pair 9F/9R (Table 1), using AmpliTaq Gold (Roche). Thermal cycling conditions were 95°C for 10 min, followed by 36 cycles of 95°C for 1 min, 64°C for 40 s, 72°C for 40 s, followed by 72°C for 10 min. After purification (QiaQuick PCR purification kit, Qiagen), the PCR product was sequenced using primers 1F-8R and 9R and the ABI BigDye Terminator Sequencing kit (Applied Biosystems), according to standard protocols.
Polymorphism genotyping
Polymorphism genotypes for the KORA population (n=702) were determined using PCR followed by primer extension reactions and MALDI-TOF mass spectrometric analysis (Sequenom, San Diego, CA, USA) using the Homogenous MassEXTEND® Assay (Sequenom). PCR was performed using primers flanking the relevant SNPs. Prior to the primer extension reaction, unincorporated primers and dNTPs were removed from the PCR product by treatment with 0.2 U Shrimp Alkaline Phosphatase (20 min at 37°C, 10 min at 85°C). Primer extension was performed by addition of a mastermix containing extension primer, dNTP/ddNTPs, and Thermosequenase (Amersham) to 7 μl dephosphorylated PCR product, followed by a thermal cycling protocol as follows: 94°C for 2 min, 55 cycles of 94°C for 5 s, 52°C for 5 s, and 72°C for 10 s. Samples were desalted by adding SpectroClean Resin. Fifteen nanoliters of sample were then spotted onto the pad of the 384-SpectroCHIP™ bioarray and subsequently subjected to mass spectrometric analysis.
Polymorphism genotyping in the remaining control populations (n=169) was performed by direct sequencing or restriction enzyme analysis according to standard protocols.
Plasmid construction
To investigate the functional consequences of DNA variants in the screened region, 0.5 μg of human genomic DNA was used as a template to amplify a fragment that contained the −2190 to +613 segment, using primer pairs 1F and 1R (Table 1). This PCR product was designated as P1 as in our previous report.23 After cloning the P1 fragment into the pGEM-T Easy vector, the plasmid was digested with NcoI and SacI and the insert was subcloned into the pGL3-Basic vector (Promega), which contains the firefly luciferase-coding sequence to generate a wild-type SCN5A promoter–luciferase fusion construct designated as pGL-P1 WT. All constructs containing mutations in the 5′ upstream region were subsequently generated using pGL-P1 WT as template. To extend the functional characterization on those DNA variants identified in exon 1 and flanking intron 1 regions, a construct encompassing the −261 to +613 segment (designated P+613 WT in our previous report23) was generated and subsequently served as template to generate mutated constructs containing variants in exon 1 and flanking intron 1.
Variants were generated by site-directed mutagenesis using the QuikChange site-directed mutagenesis kit (Stratagene) using pGL-P1 WT or pGL-P+613 WT as template, following the manufacturer's instructions. All constructs were verified by direct sequencing.
Transient transfection assays
Transient transfection analyses were performed as previously reported.23 In brief, reporter activity was assayed in neonatal mouse cardiomyocytes and in CHO cells. One-day-old B6D2 mice were anesthetized and killed. Neonatal hearts were removed and placed in 1 × PBS solution. Ventricular segments were digested by Trypsin-Versene (Biofluids Inc.) and cells were cultured in Dulbecco's modified Eagle's medium supplemented with 15% (v/v) NuSerum (Beckton Dickinson), 2.5 mM thymidine, and penicillin–streptomycin (10 U/ml and 10 mg/ml respectively) in a humidified 5% CO2 atmosphere at 37°C. Cells were allowed to attach for 48 h before being used. Chinese hamster ovary K1 (CHO-K1) cells were obtained from the American Type Culture Collection (Manassas, VA, USA) and cultured as previously described.23
The SCN5A promoter–luciferase fusion constructs (1 μg DNA) were transfected into the neonatal mouse cardiac cells using Fugene 6 (Roche), and into CHO cells using lipofectamine reagent (Invitrogen). In each experiment, the pRL-TK plasmid (0.05 μg; Promega), encoding Renilla luciferase, was co-transfected to normalize for experimental variability caused by differences in cell viability or transfection efficiency. Luminescence was measured 48 h after transfection by using the Dual-Luciferase Reporter Assay System (Promega). The pGL3-Basic (promoterless) plasmid was tested in each experiment and its activity level served as the baseline.
Statistical analysis
Allele frequencies between the (Caucasian) BS patient population and the (Caucasian) KORA control population were compared using χ2 statistical analysis. Expression activity of mutant construct was divided by that of wild type that was obtained from the same transfection experiment to generate activity relative to wild type. For each construct analyzed, data obtained was based on at least six separate transient transfection experiments. Data are presented as mean±SE. Reporter gene-expression activities were compared between wild-type and mutant sequences. Statistical significance level was examined with Student's t-test. Throughout, a value of P< 0.05 was considered as being statistically significant. All statistical analyses were conducted with SPSS software (version: SPSS 13.0).
Results
Polymorphism identification
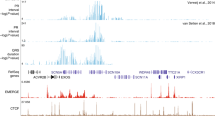
Seventeen SNPs and a haplotype consisting of two SNPs (c. [−225−1823 C>T;−225−834 T>C]) in complete linkage disequilibrium were identified in the 2.8 kb screened region (Table 2). Seven SNPs (c.−225−1790 G>A, c.−225−1657 A>G, c.−225−1294 T>G, c.−225−988 G>T, c.−225−667 A>G, c.−53+167 G>T, c.−53+274 G>A) were only identified in the control population and at low minor allele frequency (<2%). Six SNPs and the 2-SNP haplotype were identified in both case and control populations (Table 2). Among all variants identified, the most common were c.−225−2038 G>T, c.−53+114 C>T, c.−53+708 G>A, and the 2-SNP haplotype, with minor allele frequencies of >5% in at least one studied population (Table 2). Rarer alleles were often represented in a single ethnic group. For example, c.−225−1790 G>A, c.−225−1657 A>G, and the intronic variant c.−53+649 G>T were only identified in the Coriell African American control panel with minor allele frequencies of ∼1, ∼1, and ∼3% respectively. In the 5′ upstream region, 8 out of the 11 polymorphisms were identified in the −1 to −2 kb region while only three occurred within the region from the transcription start site through −1 kb. The overall frequency of the 19 variants described here and those previously reported24, 23 within the screened region was ∼1/200 base pairs. The nucleotide changes of the novel variants and variants described before were 50% transition (pyrimidine to pyrimidine or purine to purine), 46.2% transversion (purine to pyrimidine or vice versa), and 3.8% insertion/deletions. This is in agreement with previous reports indicating that transitions are more common than transversions.29
No differences in allele frequencies were detected between the (Caucasian) BS patient population and the (Caucasian) KORA control population.
In vitro functional assays
Table 3 and Figure 1 show the effects of 12 of the 17 SNPs and the haplotype identified in this study on SCN5A promoter activities. Two rare variants, c.−225−1790 G>A, and the intron 1 variant c.−53+167 G>T reduced expression activities significantly in both cardiac myocytes and non-cardiac (CHO) cells. Compared with wild type, promoter activity in c.−225−1790 G>A was 0.45±0.08 in cardiomyocytes and 0.37±0.06 in CHO cells; promoter activity of c.−53+167 G>T was 0.47±0.09 in cardiomyocytes and 0.58±0.09 in CHO cells. The c.−225−1790 G>A variant was only identified in the Coriell African American sample, and c.−53+167 G>T was identified in the Middle Tennessee control population (Table 2).

Functional analysis of DNA variants identified in the promoter and flanking regions in neonatal mouse cardiac myocytes and CHO cells. Transcriptional activities for mutant constructs are shown relative to wild type (see Methods). Firefly luciferase expression levels, which report the activities of the inserted SCN5A sequence, were normalized with the co-expressed Renilla luciferase activities to control for transfection efficiency. Data are presented as mean±SEM and each data point was derived from at least six separate transfection experiments.
Promoter activity in c.−225−775 T>A, identified in 1/176 alleles in the BS population and in Dutch and KORA controls, was significantly reduced in cardiomyocytes (0.76±0.04 relative to wild type). Promoter activity of this variant was slightly but not statistically significantly reduced in CHO cells (0.82±0.04 relative to wild type). In a similar cardiac-specific pattern, promoter activity in c.−225−1315 G>T identified in 1/176 alleles in the BS population and in KORA controls was significantly reduced in cardiomyocytes (0.55±0.07 relative to wild type). The c.−206 G>A variant, also identified in 1/176 alleles in the BS population and in Dutch and KORA controls, only reduced promoter activity in CHO cells (0.78±0.03).
About half of the characterized variants (7/12 SNPs and the haplotype) displayed activity similar to wild type in the functional assay (Table 3, Figure 1). Figure 2 summarizes the results of this and previous studies.24, 23 In vitro functional analysis of variants in this study identified five novel variants with reporter activity reduced by >20% (P<0.05) compared to wild type. The largest changes were seen with c.−225−1790 G>A. In addition, the previously reported23 variant in the core promoter region (c.−225−92 C>A) showed increased activity by >20% (P<0.05) and the six-change haplotype produced approximately 62% reduction of reporter activity in cardiomyocytes.24

Summary of SCN5A promoter polymorphisms. The diagram indicates the genomic structure of the SCN5A promoter and flanking regions, and the positions of DNA variants identified in this study. * Indicates a minor allele frequency >4% in ≥1 screened population; ^ indicates variant reporter activity <80% or >120% of wild-type control in myocytes.
Discussion
This study systematically analyzed DNA variants within the SCN5A promoter in patients with BS and a large data set of control populations. We demonstrate that DNA variants are common within the transcription regulatory regions, and that five novel variants have a significant impact on sodium channel expression in vitro.
Transcription regulatory sequences constitute only a small fraction of the roughly 95% of the human genome that does not encode proteins, but determine the level, location, and chronology of gene expression. Functional polymorphisms in transcriptional regulatory regions may affect numerous processes in mRNA biogenesis and metabolism, including processing, trafficking, stability, and translational control. Sequence variants in regulatory regions are thus logical candidates for modulating human physiology, disease susceptibility, and response to environmental stressors. DNA polymorphisms in 5′ regulatory regions have been linked to a variety of human disorders including lung cancer,30 depression,31, 32 and decreased activity of CYP3A4.33 Previous studies from our laboratory and elsewhere34 have characterized the modulating components of SCN5A core promoter in human and rat and have validated three conserved GC boxes that resemble the consensus Sp1 recognition site. Although Sp1-binding sites have been implicated as disease-associated ‘hot spots’,35, 36, 37, 38 the DNA variant screening efforts in this study did not identify any functional polymorphisms occurring in these Sp1-binding sites and the core promoter region. The absence of loss-of-function DNA variants in the core promoter of SCN5A suggests that the functional components in SCN5A core promoter are highly selectively constrained.
Changes in cis-acting response elements that lead to differences in protein abundance are thought to act by altering the binding of trans-acting proteins (transcription factors). Identifying transcription factors that bind to specific DNA regions to generate transcriptional activity in vivo remains a major challenge in this area. One approach is to analyze potential binding sites using sequence-based programs. Accordingly, we screened the variant sites identified here in SCN5A by using MatInspector, a software tool that utilizes a library of transcription factor-binding sites to predict matches in specific DNA sequences.39, 40 Interestingly, 2/15 and the two-change haplotype were predicted to alter transcription factor-binding sites (Table 3). Among them, we observed that the c.−225−1790 G>A variant, which significantly reduced expression activity, results in the generation of a GATA1-binding site and elimination of a putative ZIC2 site. ZIC2 belongs to the strong transcriptional activator Cys2–His2 family of zinc-finger transcription factors.41, 42 Elimination of this site could therefore hypothetically lead to loss of SCN5A expression. The previously reported six-change haplotype results in elimination of a predicted E2F-binding site and generation of three novel WT1 sites and two ZF5 sites, which is also consistent with the in vitro expression activity data and the loss-of-function phenotype observed in individuals carrying this haplotype allele (Table 3). Further investigation of these DNA–protein interactions predicted ‘in silico’ will be needed to validate their biological significance.
The polymorphisms we identify here may be associated with variability in basal electrocardiographic indices such as QRS duration, a manifestation of ventricular conduction velocity; this was a major effect of the six-change haplotype variant we have recently reported.24 On the other hand, such variants may be clinically silent in the basal state, but still modulate sodium-channel-related physiology in the face of environmental stressors such as transient myocardial ischemia or exposure to sodium channel-blocking drugs. Some of the variants identified here did not alter promoter activity in vitro and hence may simply be non-functional polymorphisms.
In summary, DNA polymorphisms displaying variant in vitro activity have been identified in the promoter region of the cardiac sodium channel gene: 2/88 (2.3%) BS patients and 11/1033 (1.1%) control individuals have alleles in this region of SCN5A that alter sodium channel function. The data further reinforce the concept that sodium channel function varies on a transcriptional level even among apparently normal subjects.
References
Balser JR : Inherited sodium channelopathies: models for acquired arrhythmias? Am J Physiol Heart Circ Physiol 2002; 282: H1175–H1180.
Roden DM, Balser JR, George Jr AL, Anderson ME : Cardiac ion channels. Annu Rev Physiol 2002; 64: 431–475.
George Jr AL, Varkony TA, Drabkin HA et al: Assignment of the human heart tetrodotoxin-resistant voltage-gated Na+ channel alpha-subunit gene (SCN5A) to band 3p21. Cytogenet Cell Genet 1995; 68: 67–70.
Wang Q, Shen J, Li Z et al: Cardiac sodium channel mutations in patients with long QT syndrome, an inherited cardiac arrhythmia. Hum Mol Genet 1995; 4: 1603–1607.
Tan HL, Bink-Boelkens MT, Bezzina CR et al: A sodium-channel mutation causes isolated cardiac conduction disease. Nature 2001; 409: 1043–1047.
Benson DW, Wang DW, Dyment M et al: Congenital sick sinus syndrome caused by recessive mutations in the cardiac sodium channel gene (SCN5A). J Clin Invest 2003; 112: 1019–1028.
Adler E, Fuster V : SCN5A – a mechanistic link between inherited cardiomyopathies and a predisposition to arrhythmias? JAMA 2005; 293: 491–493.
Chen Q, Kirsch GE, Zhang D et al: Genetic basis and molecular mechanism for idiopathic ventricular fibrillation. Nature 1998; 392: 293–296.
Fukuda K, Davies SS, Nakajima T et al: Oxidative mediated lipid peroxidation recapitulates proarrhythmic effects on cardiac sodium channels. Circ Res 2005; 97: 1262–1269.
Shaw RM, Rudy Y : Electrophysiologic effects of acute myocardial ischemia. A mechanistic investigation of action potential conduction and conduction failure. Circ Res 1997; 80: 124–138.
The Cardiac Arrhythmia Suppression Trial (CAST) Investigators: Preliminary report: effect of encainide and flecainide on mortality in a randomized trial of arrhythmia suppression after myocardial infarction. N Engl J Med 1989; 321: 406–412.
Coromilas J, Saltman AE, Waldecker B, Dillon SM, Wit AL : Electrophysiological effects of flecainide on anisotropic conduction and reentry in infarcted canine hearts. Circulation 1995; 91: 2245–2263.
Antzelevitch C : Cellular basis and mechanism underlying normal and abnormal myocardial repolarization and arrhythmogenesis. Ann Med 2004; 36 (Suppl 1): 5–14.
Krishnan SC, Antzelevitch C : Flecainide-induced arrhythmia in canine ventricular epicardium. Phase 2 reentry? Circulation 1993; 87: 562–572.
Lukas A, Antzelevitch C : Differences in the electrophysiological response of canine ventricular epicardium and endocardium to ischemia. Role of the transient outward current. Circulation 1993; 88: 2903–2915.
Baroudi G, Pouliot V, Denjoy I, Guicheney P, Shrier A, Chahine M : Novel mechanism for Brugada syndrome: defective surface localization of an SCN5A mutant (R1432G). Circ Res 2001; 88: E78–E83.
Roden DM : Human genomics and its impact on arrhythmias. Trends Cardiovasc Med 2004; 14: 112–116.
Alkema WB, Johansson O, Lagergren J, Wasserman WW : MSCAN: identification of functional clusters of transcription factor binding sites. Nucleic Acids Res 2004; 32: W195–W198.
Grad YH, Roth FP, Halfon MS, Church GM : Prediction of similarly acting cis-regulatory modules by subsequence profiling and comparative genomics in Drosophila melanogaster and D. pseudoobscura. Bioinformatics 2004; 20: 2738–2750.
Butler JE, Kadonaga JT : The RNA polymerase II core promoter: a key component in the regulation of gene expression. Genes Dev 2002; 16: 2583–2592.
Smale ST, Kadonaga JT : The RNA polymerase II core promoter. Annu Rev Biochem 2003; 72: 449–479.
Hoogendoorn B, Coleman SL, Guy CA et al: Functional analysis of human promoter polymorphisms. Hum Mol Genet 2003; 12: 2249–2254.
Yang P, Kupershmidt S, Roden DM : Cloning and initial characterization of the human cardiac sodium channel (SCN5A) promoter. Cardiovasc Res 2004; 61: 56–65.
Bezzina CR, Shimizu W, Yang P et al: Common sodium channel promoter haplotype in Asian subjects underlies variability in cardiac conduction. Circulation 2006; 113: 338–344.
Brugada J, Brugada R, Brugada P : Right bundle-branch block and ST-segment elevation in leads V1 through V3: a marker for sudden death in patients without demonstrable structural heart disease. Circulation 1998; 97: 457–460.
Brugada P, Brugada J : Right bundle branch block, persistent ST segment elevation and sudden cardiac death: a distinct clinical and electrocardiographic syndrome. A multicenter report. J Am Coll Cardiol 1992; 20: 1391–1396.
Pfeufer A, Jalilzadeh S, Perz S et al: Common variants in myocardial ion channel genes modify the QT interval in the general population: results from the KORA study. Circ Res 2005; 96: 693–701.
Collins FS, Brooks LD, Chakravarti A : A DNA polymorphism discovery resource for research on human genetic variation. Genome Res 1998; 8: 1229–1231.
Szathmary E : Four letters in the genetic alphabet: a frozen evolutionary optimum? Proc Biol Sci 1991; 245: 91–99.
Schenk S, Schraml P, Bendik I, Ludwig CU : A novel polymorphism in the promoter of the RAGE gene is associated with non-small cell lung cancer. Lung Cancer 2001; 32: 7–12.
Caspi A, Sugden K, Moffitt TE et al: Influence of life stress on depression: moderation by a polymorphism in the 5-HTT gene. Science 2003; 301: 386–389.
Lesch KP, Bengel D, Heils A et al: Association of anxiety-related traits with a polymorphism in the serotonin transporter gene regulatory region. Science 1996; 274: 1527–1531.
Lamba JK, Lin YS, Schuetz EG, Thummel KE : Genetic contribution to variable human CYP3A-mediated metabolism. Adv Drug Deliv Rev 2002; 54: 1271–1294.
Sheng ZH, Zhang H, Barchi RL, Kallen RG : Molecular cloning and functional analysis of the promoter of rat skeletal muscle voltage-sensitive sodium channel subtype 2 (rSkM2): evidence for muscle-specific nuclear protein binding to the core promoter. DNA Cell Biol 1994; 13: 9–23.
Cascorbi I, Henning S, Brockmoller J et al: Substantially reduced risk of cancer of the aerodigestive tract in subjects with variant – 463A of the myeloperoxidase gene. Cancer Res 2000; 60: 644–649.
Chevalier D, Cauffiez C, Bernard C et al: Characterization of new mutations in the coding sequence and 5′-untranslated region of the human prostacyclin synthase gene (CYP8A1). Hum Genet 2001; 108: 148–155.
Nakazawa F, Koyama T, Shibamiya A, Hirosawa S : Characterization of thrombomodulin gene mutations of the 5′-regulatory region. Atherosclerosis 2002; 164: 385–387.
Piedrafita FJ, Molander RB, Vansant G, Orlova EA, Pfahl M, Reynolds WF : An Alu element in the myeloperoxidase promoter contains a composite SP1-thyroid hormone-retinoic acid response element. J Biol Chem 1996; 271: 14412–14420.
Cartharius K, Frech K, Grote K et al: MatInspector and beyond: promoter analysis based on transcription factor binding sites. Bioinformatics 2005; 21: 2933–2942.
Quandt K, Frech K, Karas H, Wingender E, Werner T : MatInd and MatInspector: new fast and versatile tools for detection of consensus matches in nucleotide sequence data. Nucleic Acids Res 1995; 23: 4878–4884.
Aruga J, Nagai T, Tokuyama T et al: The mouse zic gene family. Homologues of the Drosophila pair-rule gene odd-paired. J Biol Chem 1996; 271: 1043–1047.
Benedyk MJ, Mullen JR, DiNardo S : Odd-paired: a zinc finger pair-rule protein required for the timely activation of engrailed and wingless in Drosophila embryos. Genes Dev 1994; 8: 105–117.
Acknowledgements
This work was supported by NIH Grants HL46681 and HL65962, Netherlands Heart Foundation Grants 2003B195 (CRB) and 2003T302 (AAMW), the Interuniversity Cardiology Institute of the Netherlands (project 27, AAMW), and BMBF Grants 01GI0204, 01GS0499 (SK, AP). Dr Roden is the William Stokes Professor of Experimental Therapeutics. Dr Bezzina is an Established Investigator of the Netherlands Heart Foundation (Grant 2005/T024).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Yang, P., Koopmann, T., Pfeufer, A. et al. Polymorphisms in the cardiac sodium channel promoter displaying variant in vitro expression activity. Eur J Hum Genet 16, 350–357 (2008). https://doi.org/10.1038/sj.ejhg.5201952
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.ejhg.5201952
Keywords
This article is cited by
-
Tox-Database.net: a curated resource for data describing chemical triggered in vitro cardiac ion channels inhibition
BMC Pharmacology and Toxicology (2012)
-
Brugada syndrome: Recent advances and controversies
Current Cardiology Reports (2008)
