
Abstract
Otosclerosis is a common form of hearing impairment among white adults with a prevalence of 0.3–0.4%. It is caused by abnormal bone homeostasis of the otic capsule that compromises free motion of the stapes in the oval window. Otosclerosis is in most patients a multifactorial disease, caused by both genetic and environmental factors. In some cases, the disease is inherited as a monogenic autosomal dominant trait, sometimes with reduced penetrance. However, families large enough for genetic linkage studies are extremely rare. To date, five loci (OTSC1-5) have been reported, but none of the responsible genes have been cloned yet. An additional locus, OTSC6, has been reported to the HUGO nomenclature committee but the relevant linkage study has not been published. In this study, a genome-wide linkage study was performed in a large Greek pedigree segregating autosomal dominant otosclerosis. A seventh locus, OTSC7, was localized on chromosome 6q13–16.1 with a multipoint LOD score of 7.5 in the 13.47 cM region defined by markers D6S1036 (centromeric) and D6S300 (telomeric). Linkage analysis of this new locus in 13 smaller Belgian and Dutch families has identified one family from The Netherlands in which allele segregation suggests linkage to this region. The overlap between the critical regions of these two families is a 1.06 Mb interval between the genetic markers D6S1036 (centromeric) and D6S406 (telomeric) on chromosome 6q13.
Similar content being viewed by others

Introduction
Otosclerosis is a form of progressive hearing loss due to an abnormal bone homeostasis of the otic capsule that leads to a bony fixation of the stapedial footplate in the oval window. This fixation disturbs transmission of sound waves through the middle ear ossicles, resulting in a conductive hearing impairment. In about 10% of all otosclerosis patients, an additional cochlear component develops that most likely represents the encroachment of otosclerotic foci on the labyrinthine capsule, which leads to a mixed or sensorineural hearing loss (SHL).1 Occasionally, otosclerotic damage to the cochlea produces only SHL.2
The prevalence of otosclerosis among white adults is 0.3–0.4%.3 Although its etiology is unknown, it is considered a complex disease caused by both genetic and environmental factors. Occasionally, otosclerosis is inherited as a monogenic autosomal dominant trait. However, families large enough for a genetic linkage study are rare, and in these families, factors like reduced penetrance and phenocopies complicate linkage analysis.
To date, five autosomal dominant otosclerosis loci have been reported: OTSC1 on chromosome 15q25–26,4 OTSC2 on chromosome 7q34–36,5 OTSC3 on chromosome 6p21–22,6 OTSC4 on chromosome 16q21–23.2,7 and OTSC5 on chromosome 3q22–24.8 In addition, a sixth locus, OTSC6, has been reported to the Human Genome Organisation nomenclature committee but details describing this locus have not been published. None of the disease causing genes has been identified. In this study, genome wide linkage analysis was performed in a large Greek pedigree segregating autosomal dominant otosclerosis. This analysis has identified a seventh locus, OTSC7, on chromosome 6q.
Materials and methods
Clinical diagnosis
Persons who underwent stapes microsurgery were considered affected. For the remaining patients, the clinical diagnosis of otosclerosis was based on audiological data. Pure-tone audiometry was performed on all persons with air conduction at 250, 500, 1000, 2000, 4000 and 8000 Hz, and bone conduction at 250, 500, 1000, 2000 and 4000 Hz. Otoscopy and tympanoscopy were performed to rule out outer or middle ear pathology, respectively, and stapedial reflex decay was measured to assess the mobility of the stapes.
Greek family
The family used for the genome-wide screen is a large Greek family segregating autosomal dominant otosclerosis. The family originates from a small village in the island of Rhodes. The clinical details of this family have been described recently.9 The family was expanded for this study to include 19 affecteds, seven unaffecteds, six spouses and four family members with an unknown or uncertain diagnosis. The hearing loss in this family arises in childhood around the age of 10 as a conductive hearing loss, but soon becomes mixed. The presence of an air-bone gap, closing at a frequency of 2000 Hz, together with the absence of stapedial reflexes, clinically confirmed the diagnosis of otosclerosis in these patients. In some of the family members, progression of the additional sensorineural component led to a pure SHL, masking the conductive component. The diagnosis in these latter patients was based on the family history together with the presence of additional signs, like the absence of stapedial reflexes in all patients and detailed information about the progression of the hearing loss.
All family members with hearing loss were also examined by a clinical geneticist to exclude syndromic forms of stapes fixation. In particular, movement of finger joints, shape of fingers and toes as well as facial characteristics were noted.
Dutch and Belgian families
Thirteen additional families from the Netherlands and Belgium were analyzed, and nine of these families were described previously.10 The clinical diagnosis of the four additional families was made in the same way as the other nine families.10 Persons who had stapes microsurgery were considered affected. In non-operated patients, the diagnosis was based on audiologic data, having conductive or mixed hearing loss together with absent stapedial reflexes. Because of variability in the age of onset, only persons older than 50 and with normal hearing were considered unaffected. The remaining persons were given an ‘uncertain’ affection status.
Genotype analysis
Blood samples from study participants were obtained after informed consent and genomic DNA was isolated using standard techniques.
In a large Greek family, consisting of 36 persons, a genome-wide screen was performed by deCODE genetics (Reykjavik, Iceland) using approximately 500 microsatellite markers with an average marker density of 8 cM. For fine mapping of the candidate region, we selected additional microsatellite markers from public databases.
The microsatellite markers used to analyze linkage to the OTSC7 locus in 10 Belgian and 3 Dutch families were D6S1596, D6S1589 and D6S275. Genotyping was performed on DNA from 165 persons: 83 were diagnosed as affected, 69 persons were labeled unaffected and 13 were given an uncertain diagnosis.
For the microsatellite markers we analyzed, polymerase chain reaction (PCR) amplification was carried out using standard conditions. One of the primers was synthesized with an M13 sequence at the 5′ end. A fluorescently labeled M13 primer was included in the PCR reaction, thus labeling the PCR product. Capillary electrophoresis and pattern visualization were performed using an ABI PRISM 3100 DNA sequencer (Applied Biosystems Inc., Foster City, USA). Allele sizes were determined using the GENESCAN 2.1.1. and Genotyper V3.7 software (Applied Biosystems Inc.).
Linkage analysis
Two-point LOD scores were calculated using the MLINK computer program.11 For linkage analysis, otosclerosis was coded as an autosomal dominant trait with reduced penetrance. The frequency of the otosclerosis gene was set at 0.0001 and the penetrance at 90%. To allow for possible phenocopies, this chance was set at 1%, because without surgical exploration it is often difficult to exclude hearing loss of other origin. Equal recombination frequencies between males and females were assumed. Allele frequencies were taken from the Human Genome Database (GDB)12 for the Caucasian population, except for markers D6S1960 and D6S1036, for which no data were available. For these markers, equal allele frequencies were assumed. Multipoint LOD scores were calculated using Simwalk v2.91 via the easyLINKAGE program.13 The easyLINKAGE program uses allele frequencies based on the alleles seen in the family. Haplotypes were constructed manually as well as with the program Simwalk v2.91. Both methods gave the same result. Linkage simulations were performed with the program FastSlink v2.5114 via the easyLINKAGE program.13
Mutation analysis
Primers were designed from intronic sequences flanking the exons of the genes of interest. The exon/intron structure of LOC643095 was predicted using the GeneMark.hmm,15 GenScan16 and GrailEXP17 gene prediction programs. PCR amplification was performed using genomic DNA from patients and controls. Direct sequencing of the PCR products was performed on forward and reverse strands using an ABI 3130XL sequencer with the BigDye® Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems Inc.).
Results and discussion
Genetic analysis of the Greek family
Previous analysis in the large Greek family excluded linkage to the four known OTSC loci (1–3, 5), COL1A1 and COL1A2, and no disease causing mutations were identified in the coding region of NOG.9 The family was expanded for this study to include 19 affecteds, seven unaffecteds, six spouses and four family members with an unknown or uncertain diagnosis. A genome-wide screen revealed a linked region on chromosome 6q. A maximum two-point LOD score of 4.42 was obtained for marker D6S458 at recombination fraction (θ) 0.00 (Table 1).
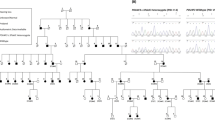
Seventeen extra markers were used to reconstruct haplotypes to confine the critical interval of the linked region. Recombinant individuals (case VI:9 proximal and case IV:8 distal) delineate a 13.47 cM candidate interval between markers D6S1036 (centromeric) and D6S300 (telomeric) on chromosome 6q13–16.1 (Figures 1 and 3). A maximum two-point LOD score of 5.32 was obtained for the marker D6S406 (Table 1) and multipoint analysis using all 21 markers genotyped, reached a maximum LOD score of 7.5.

Pedigree of the Greek family with autosomal dominant otosclerosis, showing the most likely haplotypes for the chromosome 6q13–16.1 markers. The haplotype linked to otosclerosis is indicated with a black bar. For clarity, not all analyzed markers mentioned in Table 1 are shown.

Refinement of the candidate region by identification of key recombinant individuals. The thinner part of the black line in person III:1 from the Dutch family indicates a non-informative region. Not all shown markers are analyzed in both families. The horizontal box indicates the overlapping minimal candidate region located between the markers D6S1036 and D6S406.
The OTSC7 region contains 66 genes and 46 gene predictions based on the last draft of the human genome sequence (Build 36 version 1), including a strong candidate gene COL12A1. Collagen type XII is a member of the fibril-associated collagens with interrupted triple helices (FACIT). Collagens are known to be important in syndromic and non-syndromic forms of hearing impairment and in addition, COL12A1 is expressed in the cochlea.18, 19 Mutation analysis of all 66 exons and the intron–exon boundaries of COL12A1 did not reveal any disease-causing mutation.
Analysis of additional families
Additional families (10 Belgian and three Dutch) were investigated for linkage to the OTSC7 region by analyzing three markers: D6S1596, D6S1589 and D6S275.
In one Dutch family haplotype segregation and LOD score calculations revealed suggestive linkage to OTSC7 (Table 2). In the other families, linkage to this region was excluded or unlikely by LOD score calculation and/or haplotype construction (data not shown).
Refinement of the OTSC7 region
The Dutch family consists of six affecteds, three unaffecteds and one spouse. In this family, additional markers for the OTSC7 region were analyzed defining a 4.01 cM candidate region between the markers D6S1619 (centromeric) and D6S406 (telomeric) on chromosome 6q12–14.1 (Table 2, Figure 2). The highest two-point and multipoint LOD scores for this family was 1.96 for the markers D6S1036, D6S280, D6S1596. Linkage simulation performed in this family using FastSlink13, 14 showed that the LOD score of 1.96 corresponds to the theoretical maximum LOD score for this family. The Dutch candidate region contains 16 genes and 17 gene predictions (Build 36 version 1), including a possible candidate gene COL9A1. Collagen type IX is, like collagen XII, a member of the FACIT collagens and expressed in the cochlea.18, 19 Although COL9A1 is just outside the Greek candidate region, DNA sequencing was completed for all 38 exons and intron–exon boundaries in patients from both families, but did not reveal a disease-causing mutation.

Pedigree of the Dutch family with autosomal dominant otosclerosis. The haplotype linked to otosclerosis is indicated with a black bar. The black line at person III:1 indicates a non-informative region.
Under the hypothesis that both families, the Greek and the Dutch family, have a mutation in the same gene, the responsible gene must be located between the genetic markers D6S1036 (centromeric) and D6S406 (telomeric). This combined candidate region spans 1.06 Mb and contains only eight genes, five gene predictions and two pseudogenes (Figure 3, Table 3).
Under the hypothesis that the same gene is involved in the etiology of otosclerosis in both families and taking into account that the Greek and the Dutch families do not share the same linked haplotype within the OTSC7 region, these results strongly suggest that the Greek and Dutch families are not related. Therefore, they most likely don't have the same disease causing mutation.
In the Greek pedigree, the disease haplotype is shared only by affected individuals, except for one healthy individual VI:6 (Figure 1). This finding could reflect the reduced gene penetrance that is seen in many otosclerosis families. Haplotype construction with all 21 markers analyzed showed that individual VI:11 is homozygous for 18 of these markers. Therefore, it is possible for her to be homozygous for the disease-causing mutation, although this is not necessarily the case, as the mode of inheritance for otosclerosis is autosomal dominant. The audiometrical data showed no real difference in phenotype compared with the other patients. Only the right ear was worse and showed a profound hearing loss, but asymmetric hearing loss was also seen in other patients. The Greek family originates from a small village on the island of Rhodes and therefore it is possible that the parents of individual VI:11 are related and share some of their alleles. Individual VI:5 is a daughter of a consanguineous marriage in which both parents are affected. She is homozygous only for a part of the Greek candidate region and not for the overlapping minimal candidate region. The phenotype was also comparable to the other patients.
DNA sequencing of all exons and the intron–exon boundaries of these eight genes and five gene predictions in patients from both families did not reveal a disease-causing mutation. There are numerous explanations for this negative result, including (1) the Dutch family is not linked to OTSC7 and the haplotype segregation is a coincidence. Therefore, the disease-causing gene is located elsewhere in the region defined by the Greek family; (2) the Dutch family is linked to OTSC7 and the gene is located in the overlapping minimal candidate region but the mutation could not be detected by the screening methodology we used. Examples would include deletions, large rearrangement, or mutations outside known exons. It is also possible that the OTSC7 gene is not yet annotated in genetic databases.
Linkage analysis is the first step in the identification of genes for monogenic forms of otosclerosis. It is likely that the responsible genes play a role in the bone homeostasis. Little is known about this process, and the identification of the involved genes will elucidate the associated molecular mechanisms. Understanding these mechanisms may in turn lead to new strategies for prevention and treatment of this disease.
References
Ramsay HA, Linthicum Jr FH : Mixed hearing loss in otosclerosis: indication for long-term follow-up. Am J Otol 1994; 15: 536–539.
Salvinelli F, Trivelli M, Greco F, Linthicum Jr FH : Otosclerosis and cochlear otosclerosis: a post mortem study on temporal bones. Eur Rev Med Pharmacol Sci 1999; 3: 179–182.
Declau F, Van Spaendonck M, Timmermans JP et al: Prevalence of otosclerosis in an unselected series of temporal bones. Otol Neurotol 2001; 22: 596–602.
Tomek MS, Brown MR, Mani SR et al: Localization of a gene for otosclerosis to chromosome 15q25–q26. Hum Mol Genet 1998; 7: 285–290.
Van Den Bogaert K, Govaerts PJ, Schatteman I et al: A second gene for otosclerosis, OTSC2, maps to chromosome 7q34–36. Am J Hum Genet 2001; 68: 495–500.
Chen W, Campbell CA, Green GE et al: Linkage of otosclerosis to a third locus (OTSC) on human chromosome 6p21.3–22.3. J Med Genet 2002; 39: 473–477.
Brownstein Z, Goldfarb A, Levi H, Frydman M, Avraham KB : Chromosomal mapping and phenotypic characterization of hereditary otosclerosis linked to the OTSC4 locus. Arch Otolaryngol Head Neck Surg 2006; 132: 416–424.
Van Den Bogaert K, De Leenheer EM, Chen W et al: A fifth locus for otosclerosis, OTSC5, maps to chromosome 3q22–24. J Med Genet 2004; 41: 450–453.
Iliadou V, Van Den Bogaert K, Eleftheriades N et al: Monogenic nonsyndromic otosclerosis: audiological and linkage analysis in a large Greek pedigree. Int J Pediatr Otorhinolaryngol 2006; 70: 631–637.
Van Den Bogaert K, Govaerts PJ, De Leenheer EM et al: Otosclerosis: a genetically heterogeneous disease involving at least three different genes. Bone 2002; 30: 624–630.
Lathrop GM, Lalouel JM, Julier C, Ott J : Strategies for multilocus linkage analysis in humans. Proc Natl Acad Sci USA 1984; 81: 3443–3446.
The GDB human genome database, http://www.gdb.org/.
Lindner TH, Hoffmann K : easyLINKAGE: a PERL script for easy and automated two-/multi-point linkage analyses. Bioinformatics 2005; 21: 405–407.
Weeks D, Ott G, Lathrop GM : SLINK: a general simulation program for linkage analysis. Am J Hum Genet 1990; 47: A204.
Lomsadze A, Ter-Hovhannisyan V, Chernoff YO, Borodovsky M : Gene identification in novel eukaryotic genomes by self-training algorithm. Nucleic Acids Res 2005; 33: 6494–6506.
Burge C, Karlin S : Prediction of complete gene structures in human genomic DNA. J Mol Biol 1997; 268: 78–94.
Xu Y, Uberbacher EC : Automated gene identification in large-scale genomic sequences. J Comput Biol 1997; 4: 325–338.
Robertson NG, Khetarpal U, Gutierrez-Espeleta GA, Bieber FR, Morton CC : Isolation of novel and known genes from a human fetal cochlear cDNA library using subtractive hybridization and differential screening. Genomics 1994; 23: 42–50.
Human Cochlear cDNA library and EST database, http://hearing.bwh.harvard.edu/estinfo.htm.
Acknowledgements
We thank all the family members for their cooperation. This work was supported by the European Commission FP6 Integrated project EuroHear LSHG-CT-20054-512063 and NIH grant R01DC05218. MT holds a predoctoral research position with the Institute for the Promotion of Innovation through Science and Technology in Flanders (IWT-Vlaanderen). The work in the laboratory of MBP was supported by a grant from Oticon Foundation, Denmark.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Thys, M., Van Den Bogaert, K., Iliadou, V. et al. A seventh locus for otosclerosis, OTSC7, maps to chromosome 6q13–16.1. Eur J Hum Genet 15, 362–368 (2007). https://doi.org/10.1038/sj.ejhg.5201761
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.ejhg.5201761
Keywords
This article is cited by
-
Genetics of otosclerosis: finally catching up with other complex traits?
Human Genetics (2022)
-
A pathogenic deletion in Forkhead Box L1 (FOXL1) identifies the first otosclerosis (OTSC) gene
Human Genetics (2022)
-
Insufficient evidence for a role of SERPINF1 in otosclerosis
Molecular Genetics and Genomics (2019)
-
Exclusion of TNFRSF11B as Candidate Gene for Otosclerosis in Campania Population
Indian Journal of Otolaryngology and Head & Neck Surgery (2014)
-
Controversies in RELN/reelin expression in otosclerosis
European Archives of Oto-Rhino-Laryngology (2012)
