
Abstract
Hirschsprung disease (HSCR) is characterised by intestinal obstruction resulting from an absence of ganglion cells in the intestinal tract. The mutations in the major gene, RET, associated with isolated HSCR, are dominant loss-of-function mutations with incomplete penetrance and variable expressivity. We have ascertained a large inbred Israeli-Arab family segregating HSCR. Sequencing of the RET gene showed a splicing mutation, IVS6+5G−>A, in the homozygous state in all the females with severe forms of HSCR and in the heterozygous state in the male patient with short-segment HSCR. The recently described hypomorphic-RET predisposing allele, rs2435357, was transmitted in the heterozygous state to the male patient, but was not transmitted to the three affected females. Although the heterozygous IVS6+5G−>A is of low-penetrance for short-segment HSCR disease, the homozygous state is fully penetrant for total aganglionosis or long-segment HSCR. As in other inbred populations segregating a weakly penetrant RET allele (Mennonite), our findings support the hypothesis that the penetrance of RET gene mutations for the HSCR phenotype depends on: (i) the nature of the mutation, (ii) the allele dosage and (iii) modifier-loci.
Similar content being viewed by others

Introduction
Hirschsprung disease (HSCR) is characterised by intestinal obstruction resulting from the absence of ganglion cells in a variable portion of the intestinal tract. The incidence of HSCR is 1:5000 live-births in Caucasian populations.1 Symptoms range from abdominal distension and failure to pass stools in neonates, to chronic constipation and enterocolitis in childhood. In 80% of individuals, aganglionosis is restricted to the rectosigmoid colon (short-segment disease, S-HSCR), but in ∼15% the aganglionosis extends proximal to the sigmoid colon (long-segment disease, L-HSCR). In ∼5% of individuals, aganglionosis affects the entire large intestine (total colonic aganglionosis, TCA). Total intestinal aganglionosis (TIA) extending from the duodenum to the rectum is the rarest form and is usually fatal. The RET gene, located on chromosome 10q11.21, is the major gene in nonsyndromic HSCR.2, 3
The vast majority of families with HSCR show linkage to the RET locus.4 Heterozygous mutations within the RET gene coding sequence are identified only in 50% of (linked) families and 10–20% of sporadic cases5, 6, 7, 8, 9 and are characterised by incomplete sex biased penetrance and a variable extension of aganglionosis.10, 11 The estimated penetrance is 72% in males and 51% in females.5 Recently a major role of noncoding variations in intron 1 of RET (hypomorphic alleles) has been demonstrated by several studies.9, 12, 13, 14, 15
In this report, we describe an inbred Israeli-Arab family with HSCR where a splicing mutation segregates in affected family members either in the homozygous state resulting in TCA or L-HSCR (females) or in the heterozygous state resulting in S-HSCR (male).
Patients
The patients are all members of a large consanguineous Israeli-Arab family. In one branch of the family three female siblings have TCA (individuals III-5 (deceased), III-6 and III-9, Figure 1) and one female has L-HSCR (individual III-11, Figure 1), and in the second branch of the family one male has S-HSCR (individual III-2, Figure 1). Patients III-4, III-8 and III-11 also have congenital autosomal recessive ichthyosis, which is unrelated to the HSCR. The clinical features of the patients are summarized in Table 1. The research study was reviewed and approved by the Ethics Committee (CCPPRB approval 95-05-03, AP-HP, Paris).

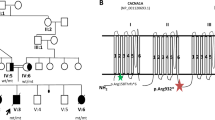
The haplotypes and mutation analysis in the affected and unaffected family members in the families with HSCR. The order of the genetic markers analysed is shown in the upper left-hand corner. The arrow indicates the proband.
Methods
DNA was isolated by standard methods. Linkage analysis to the RET gene locus was performed using polymorphic genetic markers D10S141, RET INT-5 and StCl2, followed by sequencing of the SNP rs2435357 (IVS1-C>T). Mutation screening of the coding sequence of the RET gene was performed with primers designed for exons and the flanking splice sites. PCR products were directly sequenced in both directions on an ABI PRISM 3100 DNA sequencer (Perkin Elmer-Applied Biosystems) using the Big Dye Terminator method according to the manufacturer's instructions.
Results
Linkage to the RET gene locus was established. The haplotype 165-C-184-144 segregated with the HSCR phenotype (Figure 1). A splice-mutation, IVS6+5G−>A, was identified; this was not found in 120 control chromosomes. After the complete sequencing of the RET gene, no other sequence changes were found. The mutation IVS6+5G−>A involves a known canonical splice site, where the intronic nucleotide +5 is known to participate in the donor splice site processes. When the IVS6+5G−>A mutation was tested with a programme (http://www.fruitfly.org/seq_tools/splice.html), which is designed to predict the efficiency of splice sites, the score of the wild donor site sequence decreased from one to 0.54.
All the patients with TCA and the patient with L-HSCR were homozygous for the mutation (Figure 1). The same mutation in heterozygous state was found in the boy with S-HSCR and in seven healthy family members (Figure 1). Individuals II-1, II-4 and III-2 carried the hypomorphic allele T (SNP rs2435357) in trans. The rs2435357 allele was not present in any of the females homozygous for the IVS6+5G−>A mutation.
Discussion
Heterozygous mutations of the RET proto-oncogene occur in families with TCA, L- or S-HSCR. 7, 16, 17, 18 Homozygous RET gene mutations causing HSCR are extremely rare.
Only one patient with TIA and three with TCA owing to homozygous RET mutations have been reported in the literature (Table 2). In the family described by Geneste et al,19 as in our patients, TCA was caused by a homozygous RET gene mutation, whereas in contrast to our family, L-HSCR in another set of their patients was caused by the same mutation in the heterozygous state.
In this study, we illustrate the effect of RET gene dosage on the penetrance and expressivity of the HSCR phenotype. Although the IVS6+5G−>A mutation in heterozygous state is of low penetrance for S-HSCR disease (less than 12.5% if the obligate untyped carriers in generation I are included), there is full penetrance (100%) in the homozygous state. Homozygous patients show little variability of expression (TCA with small bowel involvement and L-HSCR).
In a study describing homozygous mutation inheritance in the EDNRB in a large inbred Mennonite kindred with HSCR, most of the affected individuals were homozygous for the mutated allele, although some heterozygotes were also described.20 Homozygotes and heterozygotes for the EDNRB mutation W276C had a 74 and 21% risk, respectively, of developing HSCR. The EDNRB-mutation showed incomplete penetrance, as some unaffected individuals from this family were also found to be homozygous. In addition, some affected individuals did not carry the mutation, suggesting the presence of additional susceptibility loci contributing to HSCR inheritance.21 Even in an isolated population, such as the Mennonites, HSCR is a multigenically inherited disease involving interaction between the hypomorphic-EDNRB allele and one or more RET HSCR-susceptibility variants.22 In our study, all the family members who are homozygous for the RET gene mutation have severe forms of HSCR. None of them carries the hypomorphic allele rs2435357, which we chose because of homology and evolutionary conservation between rodents and primates and because in vitro studies have highlighted an enhancer role for this region.15, 23
Two males, II-1 and III-2, are heterozygotes for the splice mutation and the hypomorphic allele; however, individual III-2 is affected (S-HSCR), whereas his haplo-identical father, II-1, is unaffected. Additional genetic changes are thought to be responsible for the variable expressivity of the disease in the homozygous and heterozygous patients described in this study.
As suggested in other inbred populations segregating a weakly penetrant RET predisposing allele, our findings support the hypothesis that the penetrance of RET gene mutations for the HSCR phenotype depend on: (i) the nature of the mutation, (ii) the allele dosage and (iii) the modifier-loci.
The results of this study emphasise the importance of ascertaining the molecular basis of HSCR in families with more than one affected individual, especially if they originate from a small-inbred population. The detection of a RET gene mutation allows the families to be offered genetic counselling and enables early disease detection in the homozygous individuals.
References
Bodian M, Carter C : A family study of Hirschsprung disease. Ann Hum Genet 1963; 26: 261–277.
Edery P, Lyonnet S, Mulligan LM et al: Mutations of the RET proto-oncogene in Hirschsprung's disease. Nature 1994; 367: 378–380.
Romeo G, Ronchetto P, Luo Y et al: Point mutations affecting the tyrosine kinase domain of the RET proto-oncogene in Hirschsprung's disease. Nature 1994; 367: 377–378.
Bolk S, Pelet A, Hofstra RM et al: A human model for multigenic inheritance: phenotypic expression in Hirschsprung disease requires both the RET gene and a new 9q31 locus. Proc Natl Acad Sci USA 2000; 97: 268–273.
Attié T, Pelet A, Edery P et al: Diversity of RET proto-oncogene mutations in familial and sporadic Hirschsprung disease. Hum Mol Genet 1995; 4: 1381–1386.
Angrist M, Bolk S, Thiel B et al: Mutation analysis of the RET receptor tyrosine kinase in Hirschsprung disease. Hum Mol Genet 1995; 4: 821–830.
Seri M, Yin L, Barone V et al: Frequency of RET mutations in long- and short-segment Hirschsprung disease. Hum Mutat 1997; 9: 243–249.
Gabriel SB, Salomon R, Pelet A et al: Segregation at three loci explains familial and population risk in Hirschsprung disease. Nat Genet 2002; 31: 89–93.
Pelet A, de Pontual L, Clement-Ziza M et al: Homozygosity for a frequent and weakly penetrant predisposing allele at the RET locus in sporadic Hirschsprung disease. J Med Genet 2005; 42 : e18.
Lyonnet S, Bolino A, Pelet A et al: A gene for Hirschsprung disease maps to the proximal long arm of chromosome 10. Nat Genet 1993; 4: 346–350.
Angrist M, Kauffman E, Slaugenhaupt SA et al: A gene for Hirschsprung disease (megacolon) in the pericentromeric region of human chromosome 10. Nat Genet 1993; 4: 351–356.
Fitze G, Schierz M, Kuhlisch E et al: Novel intronic polymorphisms in the RET proto-oncogene and their association with Hirschsprung disease. Hum Mutat 2003; 22: 177.
Garcia Barcelo MM, Sham MH, Lui VC et al: Chinese patients with sporadic Hirschsprung's disease are predominantly represented by a single RET haplotype. J Med Genet 2003; 40: e122.
Burzynski GM, Nolte IM, Osinga J et al: Localizing a putative mutation as the major contributor to the development of sporadic Hirschsprung disease to the RET genomic sequence between the promoter region and exon 2. Eur J Hum Genet 2004; 12: 604–612.
Emison ES, McCallion AS, Kashuk CS et al: A common sex-dependent mutation in a RET enhancer underlies Hirschsprung disease risk. Nature 2005; 434 : 857–863.
Edery P, Pelet A, Mulligan LM et al: Long segment and short segment familial Hirschsprung's disease: variable clinical expression at the RET locus. J Med Genet 1994; 31: 602–606.
Inoue K, Shimotake T, Iwai N : Mutational analysis of RET/GDNF/NTN genes in children with total colonic aganglionosis with small bowel involvement. Am J Med Genet 2000; 93: 278–284.
Shimotake T, Go S, Inoue K, Tomiyama H, Iwai N : A homozygous missense mutation in the tyrosine E kinase domain of the RET proto-oncogene in an infant with total intestinal aganglionosis. Am J Gastroenterol 2001; 96: 1286–1291.
Geneste O, Bidaud C, De Vita G et al: Two distinct mutations of the RET receptor causing Hirschsprung's disease impair the binding of signalling effectors to a multifunctional docking site. Hum Mol Genet 1999; 8: 1989–1999.
Puffenberger EG, Kauffman ER, Bolk S et al: Identity-by-descent and association mapping of a recessive gene for Hirschsprung disease on human chromosome 13q22. Hum Mol Genet 1994; 3: 1217–1225.
Puffenberger EG, Hosoda K, Washington SS et al: A missense mutation of the endothelin-B receptor gene in multigenic Hirschsprung's disease. Cell 1994; 79: 1257–1266.
Carrasquillo MM, McCallion AS, Puffenberger EG, Kashuk CS, Nouri N, Chakravarti A : Genome-wide association study and mouse model identify interaction between RET and EDNRB pathways in Hirschsprung disease. Nat Genet 2002; 32: 237–244.
McCallion AS, Stames E, Conlon RA, Chakravarti A : Phenotype variation in two-locus mouse models of Hirschsprung disease: tissue-specific interaction between Ret and Ednrb. Proc Natl Acad Sci USA 2003; 100: 1826–1831.
Acknowledgements
We are grateful to the families who participated in this study. We thank Dr Gabrielle Halpern for her help with editing the manuscript and Irit Lis for preparing the figures.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Basel-Vanagaite, L., Pelet, A., Steiner, Z. et al. Allele dosage-dependent penetrance of RET proto-oncogene in an Israeli-Arab inbred family segregating Hirschsprung disease. Eur J Hum Genet 15, 242–245 (2007). https://doi.org/10.1038/sj.ejhg.5201733
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.ejhg.5201733
