
Abstract
Achondroplasia (ACH) and hypochondroplasia (HCH) are two autosomal-dominant skeletal disorders caused by recurrent missense FGFR3 mutations in the transmembrane (TM) and tyrosine kinase 1 (TK1) domains of the receptor. Although 98% of ACH cases are accounted for by a single G380R substitution in the TM, a common mutation (N540K) in the TK1 region is detected in only 60–65% of HCH cases. The aim of this study was to determine whether the frequency of mutations in patients with HCH was the result of incomplete mutation screening or genetic heterogeneity. Eighteen exons of the FGFR3 gene were entirely sequenced in a cohort of 25 HCH and one ACH patients in whom common mutations had been excluded. Seven novel missense FGFR3 mutations were identified, one causing ACH and six resulting in HCH. Six of these substitutions were located in the extracellular region and four of them creating additional cysteine residues, were associated with severe phenotypes. No mutations were detected in 19 clinically diagnosed HCH patients. Our results demonstrate that the spectrum of FGFR3 mutations causing short-limb dwarfism is wider than originally recognised and emphasise the requirement for complete screening of the FGFR3 gene if appropriate genetic counselling is to be offered to patients with HCH or ACH lacking the most common mutations and their families.
Similar content being viewed by others

Introduction
Achondroplasia (ACH, MIM# 100800) and hypochondroplasia (HCH, MIM# 146000) are two common autosomal-dominant skeletal dysplasias characterised by short stature. Both conditions share clinical and radiological features including macrocephaly, brachydactyly, metaphyseal flaring, shortening of the pedicles of the vertebrae, narrowing of interpediculate distance, square iliae and short femoral necks. However, the abnormalities seen in HCH are less severe than those seen in ACH. In some patients, the clinical features are very subtle making the diagnosis of HCH difficult in those cases.1, 2
Genetically, those diseases are allelic and have been shown to be associated with recurrent mutations in the FGFR3 gene encoding one member of the FGFR subfamily of tyrosine kinase (TK) receptors.3, 4, 5 The four FGFR (1–4) members share a common organisation comprising three extracellular immunoglobulin-like loops (Ig I–III), one hydrophobic transmembrane (TM) domain and two cytoplasmic TK sub-domains TK1 and TK2 responsible for the catalytic activity.6
Although more than 98% of ACH cases harbour a recurrent G380R substitution in the TM domain of the receptor, HCH is mostly accounted for by a common N540K amino-acid change in the TK1 domain. Only 60–65% of clinically diagnosed HCH patients carry that mutation. However, additional substitutions at positions 328 (N328I), 538 (I538V), 540 (N540S,T) and 650 (K650N,Q) have been reported,7, 8, 9, 10 but only account for a few cases, raising the question of possible genetic heterogeneity in HCH.11, 12, 13, 14 In order to assess this possibility, a new set of intronic primers were designed and used for sequencing 18 exons of the FGFR3 gene in a series of 25 HCH and one ACH patients not carrying the common pathogenic mutations. Seven novel FGFR3 mutations were identified in the extracellular or TM domain. Amino-acid substitutions creating cysteine residues were associated with severe phenotypes. Our results widen the spectrum of FGFR3 mutations causing HCH and ACH.
Patients and methods
A series of 75 patients including 10 familial cases and 65 sporadic forms were clinically diagnosed as having HCH or ACH based on clinical and radiological examination by at least one of the authors (Table 1).
Blood samples were collected after informed consent from all these patients. Parental blood samples were also obtained and used for sequencing when a de novo sporadic mutation had been identified in a patient. Genomic DNA was extracted from lymphocytes by using standard procedures. A set of intronic primers was designed based on the genomic sequence of the human FGFR3 gene (accession number: AY 768549) and used to amplify exons 2–19 (primer sequences and PCR conditions are available on request). PCR products were directly sequenced using the Big Dye Sequencing kit (Applied Biosystem, Foster City, CA, USA) on an ABI 3100 automatic DNA sequencer.
Radiographs from patients carrying novel FGFR3 mutations were re-examined after mutation detection. The fibula:tibia length ratio was calculated according to the method of Matsui et al.2
Informed consent for DNA analyses was obtained from patients, their parents or both.
Results
Mutation analysis
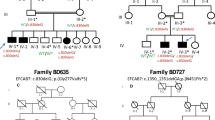
Mutations affecting codons 540 and 538 of FGFR3 were first sought in our cohort of 74 clinically diagnosed HCH cases (Table 1). Forty-seven patients (63.5%) were found to carry the N540K amino-acid change, but none of the remaining patients harboured the N540S or I538V substitutions. The previously reported K650Q and N328I mutations were detected in two cases. Interestingly, although the patient carrying the N328I substitution exhibited a typical HCH phenotype, the one carrying the K650Q mutation had a moderate phenotype (not shown) similar to previously reported individuals harbouring K650N or K650Q substitutions.9 The remaining 25 HCH patients and one individual with clinical signs of ACH but lacking the G380R or G375C mutations in exon 10, were submitted to a full screen of the whole FGFR3 coding sequence including intron–exon junctions. In this series of 26 cases, seven novel missense mutations were identified (Table 2). The patient with ACH carried a single base substitution converting serine 279 into cysteine (S279C) in the first half of the third Ig loop (Figure 1a). An adjacent mutation at position 278 (Y278C) was associated with a severe form of HCH. Two other mutations creating cysteine residues in the extracellular domain (G268C and R200C) were found in two HCH patients. One mutation (N262H) affected an asparagine residue, part of a putative glycosylation site (N–Q–T), and another occurred in the TM domain at position 381 (V381E) adjacent to the G380R mutation causing ACH (Figure 1a). None of these heterozygous substitutions were detected in DNA from unaffected parents or in 70 unrelated controls, thus confirming their de novo origin. The S84L substitution in the first Ig loop was identified in a familial form of HCH and segregated with the disease (Figure 2). During the course of this screening, several common and uncommon polymorphisms were detected in both exons and introns (Table 3). Some in the coding regions were synonymous, but we also found two non-synonymous substitutions in two HCH patients. One converted proline 449 into serine (P449S) in exon 11 and was detected both in the patient and his non-affected father. The second converted an aspartic acid residue into asparagine at position 580 (D580N) in exon 14. Parents were not available for testing. This amino-acid change affecting a non-conserved residue in a poorly conserved domain is likely to represent an uncommon variant.

Schematic representation of the receptor- and structure-based sequence alignment. (a) Distribution of novel FGFR3 mutations identified in this study. Mutations causing HCH are shown in green and the mutation causing ACH is shown in red. The different domains of the receptor are indicated: Ig I–III=immunoglobulin-like domains; A=acid box; TM=transmembrane domain ; TK1, 2=Tyrosine kinase sub-domains. Numbers at the extremities of Ig loops represent conserved cysteine residues involved in the formation of disulfide bonds. (b) Amino-acid alignment around the sites of mutation in the linker region between the Ig II and Ig III loops and the first half of the Ig III loop. Sequence identities of the three other FGFRs with FGFR3 are indicated by a dot. Mutated amino acids are red. Rectangles on top of the sequence delimit regions of β-sheet secondary structure as determined by Olsen et al.20 The putative glycosylation site is underlined.

Pedigree of the familial form of HCH (patient 6) and automated DNA sequencing of exon 3 showing the presence of a heterozygous FGFR3 mutation. Note segregation of the c250t/S84L substitution with the disease and its absence in unaffected individuals whose DNA were available (*).
Clinical and radiological data
All seven patients carrying novel FGFR3 mutations presented with short stature and clinical and radiographic anomalies of variable severity. When compared with age-matched children with ACH carrying the G380R mutation, the height of patient 1 (S279C) was −1 SD and his head circumference was 50th centile. Radiographic examination of the skeleton showed the typical features of ACH, including bowing of the radius and ulna, marked metaphyseal flaring of long bones and short femoral necks (Figure 3a–e). However, the child had epilepsy and moderate learning difficulties. An EEG revealed a temporal lobe focus and an MRI scan showed minor changes in the temporal lobe. He developed a severe kyphoscoliotic deformity affecting the lower thoracic and upper lumbar spine requiring surgical correction at the age of 7 years (Figure 3f and g). This was complicated by postoperative lower limb paralysis requiring decompressive surgery.

Radiographs of ACH patient 1 at 5 years and 9 months. (a) Short humerus with metaphyseal flaring and prominent deltoid muscle attachment. (b) Short radius and ulna with bowing of radius and metaphyseal flaring. (c) Short tibia and fibula with metaphyseal flaring and mildly increased fibular length. (d) Generalised brachydactyly with bullet shaped proximal and middle phalanges. (e) Square iliac wings with narrow sciatic notches. Short femoral necks with coxa valga deformity. (f) Kyphoscoliosis with narrow interpediculate distance. (g) Marked kyphosis with wedging of vertebrae and short pedicles.
Clinical features of patients with HCH were as follows: patient 2 (Y278C) had an ACH phenotype at birth, at the age of 6 months and during the two first years of life, with rhizomelic dwarfism, macrocephaly, midface hypoplasia with flat nasal bridge (Figure 4a), thoracolumbar kyphosis, short trident hands and mild hypotonia. X-rays at 4 weeks documented square iliac bones and oval radiolucent areas in the proximal femora (Figure 5a). At the age of 3.5 years, the phenotype had changed to typical HCH with normal craniofacial features, short stature with relatively short upper arms and thighs and lumbar hyperlordosis (Figure 4b and c). Her height was 85 cm (−3 SD). X-rays documented a low and broad pelvic configuration owing to squared iliae and short femoral necks in valgus position (Figure 5b), short and broad tibiae, disproportionately long fibulae (Figure 5c), short pedicles (Figure 5d) and hands with short middle phalanges. Patient 3 (G268C) was first examined at 18 months. Radiographs revealed marked metaphyseal flaring of the femora and tibiae and disproportionately long fibulae (Figure 5e). The patient is now 30 years and her adult size is 135 cm (−4 SD). Recent X-rays illustrate extremely narrow lumbar pedicles. Femoral necks look moderately short. There is slight reduction of metacarpal length (Figure 5f and g).

Clinical features of patient 2. (a) Patient 2 at the age of 6 months presenting as ACH with rhizomelic dwarfism, macrocephaly with midface hypoplasia, thoracolumbar kyphosis, short trident hands and mild hypotonia. (b and c) At the age of 3.5 years, an HCH phenotype has developed with normal craniofacial features (b), small stature with relatively short upper arms and thighs and lumbar hyperlordosis (c).

Radiographs of HCH patients. (a) Pelvis of patient 2 at 4 weeks with oval radiolucent area in the proximal femora. (b) Pelvis at 3.5 years. Femoral necks are short and in valgus position. (c) Short tibia and disproportionately long fibula in patient 2 at 3.5 years. (d) Lateral view of the spine with rounded vertebral bodies and dorsal scalloping in the lumbar region in patient 2 at 3.5 years. (e) Lower limbs of patient 3 at 18 months illustrating exaggerated flaring of femoral and tibial metaphyses. (f) Short hand with reduction of metacarpal bones in patient 3 at 30 years. (g) Lateral spine view showing short lumbar pedicles in patient 3 at 30 years. (h) Skeleton of patient 4 at birth. Internally truncated femoral metaphyses are noticeable. (i) Short hand of patient 4 at 6 months with mild metaphyseal flaring. (j) Short tibiae and femora with marked metaphyseal flaring in patient 4 at 3 years. (k) Left femur of patient 5 at 5 years showing moderate flaring of the distal metaphysis. (l) Disproportionately long fibula of patient 5 at 5 years. (m) Lateral view of the spine showing short lumbar pedicles in patient 5 at 12 years. (n) Pelvis of patient 6 at 3 years showing short and wide femoral necks. (o) Short lumbar pedicles are seen on the lateral spine view of the affected mother of patient 6. (p) Femurs of patient 7 at 4 years are slightly bowed. (q) Flaring of the tibial metaphyses is mild but the fibulae are longer than the tibiae in patient 7 at 4 years. (r) Mild metaphyseal cupping of metacarpals is visible in patient 7 at 8 years.
Patient 4 (R200C) had both normal length (50 cm) and normal head circumference (34.8 cm) at birth; however, mild brachycephaly was visible with low set ears and flat nasal bridge. Radiographic examination revealed short femora and tibiae with radiolucent areas in the proximal part of the femora (Figure 5h). Hands were short and stubby. Shortness of the phalanges and metacarpal bones was noticeable (Figure 5i). At 4 years, the patient's height was 93.5 cm (−2 SD) with marked bowing of the lower limbs. He had no macrocephaly (OFC 51 cm) but exhibited depressed nasal bridge. Radiographs documented metaphyseal flaring of the femora and tibiae with disproportionately long fibulae (Figure 5j). The fibula to tibia ratio (F/T) was 1.11 vs 1.01±0.02 in controls. Lumbar pedicles were relatively short. This set of features is compatible with moderately severe HCH.
Patient 5 (N262H) had a normal length at birth (54 cm) with no macrocephaly. At 5 years, radiographic examination showed short femora and tibiae and disproportionately long fibulae (F/T=1.05). Femoral heads were enlarged with shortened femoral necks. Metaphyseal flaring of the femora and tibiae was clearly visible (Figure 5k and l). At 12 years, radiographic examination of the spine revealed short lumbar pedicles (Figure 3m). Hands were stubby with short metacarpal bones and phalanges. The patient is now 16 years, his height is 146 cm (−3.5 SD) and moderate scoliosis is present.
Both patient 6 (S84L) and 7 (V381E) present with moderate HCH phenotypes. The length of patient 6 at birth was 48 cm with a head circumference of 33 cm. At 3 years, her height was 88 cm (−2 SD). The adult sizes of her affected mother and grandfather were 137 and 153 cm, respectively. Radiographs of the proband at 3 years showed wide and short femoral heads (Figure 5n). The interpedicular distance was in the normal range. Radiographs of the mother's spine showed short lumbar pedicles and femoral necks and square iliac wings (Figure 5o). Hand X-rays were normal.
Patient 7 had normal length at birth (52.5 cm). At the age of 16 months, macrocephaly was observed with flattening of the skull base. At 4 years, mild bowing of femoral diaphyses and moderate flaring of femoral and tibial metaphyses were visible on X-rays (Figure 5p and q). The fibulae were slightly longer than the tibiae (F/T=1.06), whereas vertebral bodies and hands were normal. At 8 years, his height was 116 cm (−2 SD), femoral necks were broad, there was mild metaphyseal cupping in the metacarpals (Figure 5r) and interpedicular distances were narrowed.
No intellectual disability was noted in any of the six HCH patients who all presented appropriate developmental progress.
Discussion
Until recently, the spectrum of FGFR3 mutations causing ACH and HCH was thought to be extremely limited. This was based on the observations that (i) 60–65% of clinically diagnosed HCH patients harbour the N540K/S mutation and (ii) at least 98% of ACH patients carry the same G380R mutation, irrespective of their ethnic origin, identifying nucleotide 1138 of the FGFR3 gene as the most sensitive point for germline mutation in the entire human genome.15 Our study indicates that mutations outside the TM and TK1 domains and creating cysteine residues in the extracellular region can also produce ACH and severe HCH. FGFR3 mutations affecting the extracellular domain have been previously identified in thanatophoric dysplasia (TD I) patients,16, 17 and in Muenke syndrome, a mild form of craniosynostosis originally described as non-syndromic.18 Likewise, FGFR2 mutations causing various forms of syndromic craniosynostosis (Crouzon, Apert, Pfeiffer syndromes, etc…) are found preferentially in the extracellular domain of the receptor. Interestingly, the highly conserved tyrosine residue at position 278 in FGFR3 and 281 in FGFR2, when converted into cysteine causes severe HCH (FGFR3) or Crouzon syndrome (FGFR2).19 The critical role of this amino acid was further supported by three-dimensional analysis of FGFR3c-FGF1 crystals. In this model, it was shown to directly interact with the FGF1 ligand and to participate in the formation of the βB′ strand.20 Similarly, the adjacent serine residue at position 279 in FGFR3 is also directly involved in the binding of FGF1 (Figure 1b) but belongs to the βB′-βC loop. Two other mutations creating cysteine residues (R200C and G268C) also affected highly conserved residues present in the four FGFRs. Experiments using FGFR3 constructs reproducing TD I mutations and Western blot analysis of mutant proteins under non-reducing conditions have convincingly demonstrated the presence of disulphide-bonded dimers in transfected cells.21, 22 Based on these data, we assume that formation of a disulphide bond between two mutant receptors would induce constitutive activation of the dimer resulting in ACH or severe HCH phenotypes.
Asparagine 262 is conserved between the different members of the FGFR family and is part of a putative glycosylation site N–T–Q. Its conversion into histidine is likely to disrupt the glycosylation site. Correct glycosylation of the receptor is required for its normal processing to the plasma membrane and occurs both in the endoplasmic reticulum (ER) and the Golgi system.23, 24 Defective glycosylation could impair receptor processing to the plasma membrane, by inducing transient intracellular retention of the mutant proteins in the ER or the Golgi, similar to what has been reported with the K650N mutation causing HCH.24 Another mutation (N328I) affecting a putative glycosylation site has been reported previously10 and identified in one of our patients; both mutations were associated with typical HCH phenotype in the affected individuals.
Amino acids forming the Ig I loop are poorly conserved among the different members of the FGFR family. Hence, this loop was shown to be dispensable for ligand binding and affinity as a result of its disordered three-dimensional structure.20 However, an autoregulatory role in FGFR1 function has been suggested,25 and recent surface plasmon resonance data on the FGF receptor 3 in complex with FGF1 indicate that the Ig I loop could act cooperatively with the acid box (a contiguous stretch of four acidic amino acids) to negatively regulate FGFR3 function.20
Although the precise impact of the S84L mutation on receptor function remains to be biochemically assessed, we speculate that this substitution could alter the regulatory role of the Ig I loop, thus increasing the ligand affinity of the mutant receptor. Interestingly, a S57L mutation in the FGFR2 gene has been identified in a syndromic form of craniosynostosis.19
Mutation V381E is adjacent to the G380R ACH mutation but results in a milder phenotype. An equivalent mutation V664E has been previously described in the TM domain of the Neu receptor, inducing its conversion into an oncogene through the formation of an hydrogen bond between two mutant receptors.26 Given that valine residue 381 in FGFR3 occupies the same position as valine 664 in Neu and that both are converted into glutamic acid, it is tempting to conclude that this FGFR3 mutation leads to constitutive receptor activation.27 The reduced severity of the phenotype when compared to that produced by the adjacent ACH mutation, would suggest that position 381 is less critical for receptor activation than residue 380 or that the strength of the hydrogen bond is lower.
Thorough clinical and radiological examination of patients carrying novel mutations indicated that mutations creating cysteine residues produced relatively severe phenotypes, one patient showing typical features of ACH and another exhibiting an ACH phenotype in early childhood that evolved into HCH. By contrast, the three patients carrying missense FGFR3 mutations not creating cysteine residues and the 19 HCH patients in whom no FGFR3 mutation was detectable, were phenotypically undistinguishable from patients carrying the N540K substitution. This indicates that the likelihood of finding novel FGFR3 mutations in patients showing overlapping phenotypes between ACH and HCH is much higher than in patients with classical HCH, thus emphasising the future need for complete FGFR3 analysis in patients with obvious radiographic ACH/HCH features. An appropriate strategy would be to analyse FGFR3 hot spots first, and in case of negative results, proceed to sequencing whole exons. The information obtained may be relevant for both diagnosis and genetic counselling, for example, in the case of mutation Y278C, evolution of an ACH phenotype, at birth, towards a milder HCH phenotype might be predicted. Support for this assertion was provided through our recent identification of a second patient carrying the Y278C mutation and exhibiting an ACH phenotype at birth (not shown).
Finally, our data re-emphasise the possibility of genetic heterogeneity in HCH. Unfortunately, attempts to identify a second locus have been unsuccessful so far.
References
Hall BD, Spranger J : Hypochondroplasia: clinical and radiological aspects in 39 cases. Radiology 1979; 133: 95–100.
Matsui Y, Yasui N, Kimura T et al: Genotype phenotype correlation in achondroplasia and hypochondroplasia. J Bone Joint Surg (British) 1998; 80B: 1052–1056.
Rousseau F, Bonaventure J, Legeai-Mallet L et al: Mutations in the gene encoding fibroblast growth factor receptor 3 in achondroplasia. Nature 1994; 371: 252–254.
Shiang R, Thompson LM, Zhu YZ et al: Mutations in the transmembrane domain of FGFR3 cause the most common genetic form of dwarfism, achondroplasia. Cell 1994; 78: 335–342.
Bellus G, McIntosh I, Smith EA et al: A recurrent mutation in the tyrosine kinase domain of fibroblast growth factor receptor 3 causes hypochondroplasia. Nat Genet 1995; 10: 357–359.
Schlessinger J : Cell signaling by receptor tyrosine kinases. Cell 2000; 103: 211–225.
Grigelioniené G, Hagenas L, Eklof O et al: A novel missense mutation Ile538Val in the fibroblast growth factor receptor 3 in hypochondroplasia. Hum Mut 1998; 11: 333.
Thauvin-Robinet C, Faivre L, Lewin P et al: Hypochondroplasia and stature within normal limits: another family with an Asn540Ser mutation in the fibroblast growth factor receptor 3 gene. Am J Med Genet 2003; 119A: 81–84.
Bellus G, Spector EB, Speiser P et al: Distinct missense mutations of the FGFR3 Lys 650 codon modulate receptor kinase activation and the severity of the skeletal dysplasia. Am J Hum Genet 2000; 67: 1411–1421.
Winterpacht A, Hilbert K, Stelzer C et al: A novel mutation in FGFR3 disrupts a putative N-glycosylation site and results in hypochondroplasia. Physiol Genomics 2000; 2: 9–12.
Rousseau F, Bonaventure J, Mallet L et al: Clinical and genetic heterogeneity of hypochondroplasia. J Med Genet 1996; 33: 749–752.
Stoilov I, Kilpatrick MW, Tsipouras P : A common FGFR3 gene mutation is present in achondroplasia but not in hypochondroplasia. Am J Med Genet 1995; 55: 127–133.
Grigelioniené G, Eklof O, Laurencikas E et al: Asn540Lys mutation in fibroblast growth factor receptor 3 and phenotype in hypochondroplasia. Acta Paediatr 2000; 89: 1072–1076.
Vajo Z, Francomano CA, Wilkin DJ : The molecular and genetic basis of fibroblast growth factor receptor 3 disorders: the achondroplasia family of skeletal dsyplasias, Muenke craniosynostosis and Crouzon syndrome with Acanthosis Nigricans. Endocr Rev. 2000; 21: 23–39.
Wilkie AOM : Bad bones, absent smell, selfish testes: the pleitropic consequences of human FGF receptor mutations. Cytokine Growth Factor Rev 2005; 16: 187–203.
Tavormina PL, Shiang R, Thompson L et al: Thanatophoric dysplasia (types I and II) caused by distinct mutations in fibroblast growth factor receptor 3. Nat Genet 1995; 9: 321–328.
Rousseau F, El Ghouzzi V, Delezoide A-L et al: Missense FGFR-3 mutations create cysteine residues in thanatophoric dwarfism type I (TD 1). Hum Mol Genet 1996; 5: 509–512.
Bellus G, Gaudenz K, Zackai EH et al: Identical mutations in three different fibroblast growth factor receptor genes in autosomal dominant craniosynostosis syndromes. Nat Genet 1996; 14: 174–176.
Kan S-H, Elanko N, Johnson D et al: Genomic screening of fibroblast growth factor receptor 2 reveals a wide spectrum of mutations in patients with syndromic craniosynostosis. Am J Hum Genet 2002; 70: 472–486.
Olsen SK, Ibrahimi OA, Raucci A et al: Insights into the molecular basis for fibroblast growth factor receptor autoinhibition and ligand-binding promiscuity. Proc Natl Acad Sci USA 2004; 101: 935–940.
Adar R, Monsonego-Ornan E, David P, Yayon A : Differential activation of cysteine-substitution mutants of fibroblast growth factor receptor 3 is determined by cysteine localization. J Bone Min Res 2002; 17: 860–868.
Naski MC, Wang Q, Xu J, Ornitz D : Graded activation of fibroblast growth factor receptor 3 by mutations causing achondroplasia and thanatophoric dysplasia. Nat Genet 1996; 13: 233–237.
Lievens PM-J, Liboi E : The thanatophoric dysplasia type II mutation hampers complete maturation of FGF receptor 3 which activates STAT1 from the endoplasmic reticulum. J Biol Chem 2003; 278: 17344–17349.
Lievens PM-J, Mutinelli C, Baynes D, Liboi E : The kinase activity of fibroblast growth factor receptor 3 with activation loop mutations affects receptor trafficking and signaling. J Biol Chem 2004; 279: 43254–43260.
Wang F, Kan M, Yan G et al: Alternatively spliced NH2-terminal immunoglobulin-like loop I in the ectodomain of the fibroblast growth factor (FGF) receptor 1 lowers affinity for both heparin and FGF-1. J Biol Chem 1995; 270: 10231–10235.
Sternberg MJE, Gullick WJ : Neu receptor dimerization. Nature 1989; 339: 587.
Sternberg MJE, Gullick WJ : A sequence motif in the transmembrane region of growth factor receptors with tyrosine kinase activity mediates dimerization. Protein Eng 1990; 3: 245–248.
Acknowledgements
We thank the patients and their families for their participation in this study.
This work was supported by a grant QLG1-CT-2001-02188 from the European Skeletal Dysplasia Network (ESDN).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Heuertz, S., Le Merrer, M., Zabel, B. et al. Novel FGFR3 mutations creating cysteine residues in the extracellular domain of the receptor cause achondroplasia or severe forms of hypochondroplasia. Eur J Hum Genet 14, 1240–1247 (2006). https://doi.org/10.1038/sj.ejhg.5201700
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.ejhg.5201700
Keywords
This article is cited by
-
Review of published 467 achondroplasia patients: clinical and mutational spectrum
Orphanet Journal of Rare Diseases (2024)
-
Heterozygous variant in FGFR3 underlying severe phenotypes in the second trimester: a case report
BMC Medical Genomics (2023)
-
Growth Topics in FGFR3-Related Skeletal Dysplasias
Current Treatment Options in Pediatrics (2021)
-
Achondroplasia: a comprehensive clinical review
Orphanet Journal of Rare Diseases (2019)
-
A novel S269C mutation in fibroblast growth factor receptor 3 in a Japanese child with hypochondroplasia
Human Genome Variation (2018)
