
Abstract
Sotos syndrome is an autosomal dominant condition characterised by a distinctive facial appearance, learning disability and overgrowth resulting in tall stature and macrocephaly. In 2002, Sotos syndrome was shown to be caused by mutations and deletions of NSD1, which encodes a histone methyltransferase implicated in chromatin regulation. More recently, the NSD1 mutational spectrum has been defined, the phenotype of Sotos syndrome clarified and diagnostic and management guidelines developed.
Similar content being viewed by others


In brief
-
Sotos syndrome is characterised by a distinctive facial appearance, learning disability and childhood overgrowth.
-
Sotos syndrome is associated with cardiac anomalies, renal anomalies, seizures and/or scoliosis in ∼25% of cases and a broad variety of additional features occur less frequently.
-
NSD1 abnormalities, such as truncating mutations, missense mutations in functional domains, partial gene deletions and 5q35 microdeletions encompassing NSD1, are identifiable in the majority (>90%) of Sotos syndrome cases.
-
The phenotype is largely independent of the underlying NSD1 defect, although cases with 5q35 microdeletions tend to have more severe learning disability and less pronounced overgrowth.
-
NSD1 is a histone methyltransferase that acts at H4 K20 and H3 K36.
-
NSD1 has multiple functional domains and may have complex roles in transcriptional regulation.
Introduction
Sotos syndrome was first described in 1964 by Juan Sotos and the major diagnostic criteria of a distinctive facial appearance, childhood overgrowth and learning disability were established in 1994 by Cole and Hughes.1, 2 In 2002, cloning of the breakpoints of a de novo t(5;8)(q35;q24.1) translocation in a child with Sotos syndrome led to the discovery that Sotos syndrome is caused by haploinsufficiency of the Nuclear receptor Set Domain containing protein 1 gene, NSD1.3 Subsequently, extensive analyses of overgrowth cases have shown that intragenic NSD1 mutations and 5q35 microdeletions encompassing NSD1 cause >90% of Sotos syndrome cases.4, 5, 6, 7, 8, 9, 10 In addition, NSD1 abnormalities are only very rarely identified in other overgrowth phenotypes.10 Thus, identification of an NSD1 abnormality is essentially diagnostic of Sotos syndrome and provides an objective method of identifying a condition that can be challenging to confidently diagnose clinically. Over the last few years, large-scale analyses of such molecularly confirmed cases of Sotos syndrome has clarified the clinical and molecular spectrum of the condition and provided the basis for diagnostic and management protocols (Figure 1).

Diagnostic strategy for individuals in which a diagnosis of Sotos syndrome is suspected.
Clinical overview
Cardinal features of Sotos syndrome
Three features have been designated the cardinal features of Sotos syndrome, a characteristic facial appearance, learning disability and overgrowth resulting in tall stature and macrocephaly which is evident from birth. These features are each present in over 90% of individuals with Sotos syndrome (Table 1).10
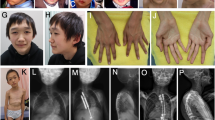
The facial appearance is most distinctive between 1 and 6 years and consists of a high, broad forehead (the head is said to resemble an inverted pear), fronto-temporal hair sparsity, malar flushing, down-slanting palpebral fissures and a pointed chin (Figure 2a). In some children, there may be atypical features such as up-slanting palpebral fissures or a normal hairline. In adulthood, the appearance remains distinctive but the face is often longer and the chin more prominent (Figure 2b).

The classic Sotos gestalt in (a) childhood and (b) adulthood.
The great majority of individuals with Sotos syndrome have some degree of learning disability. Most individuals have mild–moderate intellectual impairment but the degree of impairment is extremely broad, ranging from occasional individuals with normal development to children with profound learning difficulties requiring life-long care.
Many babies with Sotos syndrome have a birth length >2SD above the mean. The birth weight is often not proportionately increased and so babies with Sotos syndrome are usually long and thin. Before puberty the majority of children with Sotos syndrome have height and head circumference >2SD above the mean. There is often some ‘normalisation’ of height post-puberty so that the height of adults with Sotos syndrome may not be significantly above the norm. By contrast significant macrocephaly is typical in both children and adults with Sotos syndrome.
Major features of Sotos syndrome
Various medical conditions are described in at least 15% of Sotos syndrome cases and are considered major features of the condition (Table 1).10
Advanced bone age occurs in ∼75% of affected children. However, it should be noted that bone age is influenced by age at assessment and variability in interpretation.2, 10
Many individuals with Sotos syndrome have abnormalities on cranial imaging. These are largely nonspecific and no particular abnormality or pattern of abnormalities is specific to Sotos syndrome.11 Ventricular dilatation is the most commonly reported feature.10
In the neonatal period, ∼70% of babies with Sotos syndrome develop jaundice and/or have difficulty with feeding. The latter is in part because neonatal hypotonia is also very common. These neonatal problems are generally self-limiting and do not result in long-term problems.10
Cardiac and renal anomalies, seizures and scoliosis occur in 15–30% of cases and are very variable in type and severity. The cardiac anomalies range from single, self-limiting anomalies such as ASD to complex anomalies requiring interventional surgery. The most common renal abnormality is vesico-ureteric reflux, but anatomical abnormalities such as duplex kidney, absent kidney, urethral stenosis and pelvi-ureteric junction obstruction are also recognised. Infantile spasms, absence, tonic/clonic and myoclonic seizures are all described in Sotos syndrome. Scoliosis is similarly variable in severity ranging from mild, requiring no intervention, to severe, where bracing or surgery is indicated.10, 12
Other features associated with Sotos syndrome
Many other clinical features have been reported in individuals with Sotos syndrome. Some of these are quite common, such as behavioural problems and constipation, and it is possible that future clinical reviews will show that these or other features occur in >15% of cases and can be considered major features of the condition.
For the many clinical features that have been reported in only one, or a few, individuals it can be difficult to determine whether they signify genuine associations of Sotos syndrome or are coincidental findings (Table 2). It is very likely that further, currently unrecognised, features will be reported in individuals with Sotos syndrome in the future.
Differential diagnosis of Sotos syndrome
Phenotypic overlap exists between Sotos syndrome and several other clinical conditions (Table 3). In our experience, the four conditions most commonly confused with Sotos syndrome are Weaver syndrome, Bannayan–Riley–Ruvalcaba syndrome, Beckwith–Wiedemann syndrome and benign familial macrocephaly.
The greatest phenotypic overlap is between Sotos and Weaver syndromes. The facial features of these conditions are similar, particularly in infancy (Table 3). As children get older the conditions are easier to differentiate as classic Weaver syndrome cases have widely spaced palpebral fissures and a rounder face compared to the normally spaced eyes and prominent chin characteristic of Sotos syndrome.2, 10, 13 The two syndromes can also usually be differentiated molecularly as, in our experience, no classic case of Weaver syndrome has an NSD1 abnormality and we assume that Weaver syndrome is due to a different gene. Our practise is to undertake NSD1 analysis in all children in whom a diagnosis of Sotos syndrome or Weaver syndrome is being considered. If a mutation is identified the child is managed as other NSD1-positive individuals and a diagnosis of Sotos syndrome is given. Identification of the Weaver syndrome gene will facilitate discrimination between these two conditions.
Bannayan–Riley–Ruvalcaba syndrome is due to PTEN mutations in ∼60% of cases14 and, in common with Sotos syndrome, is often associated with learning disability and macrocephaly and may be associated with tall stature. Boys with Bannayan–Riley–Ruvulcaba usually have penile freckling which has not been reported in Sotos syndrome. In later childhood and adulthood lipomatosis, haemangiomatosis and features of Cowden syndrome may occur.15, 16, 17 In families with inherited PTEN mutations, there is often a family history of macrocephaly and learning difficulties that is unusual in Sotos syndrome. Molecular analysis of NSD1/PTEN should help differentiate the two conditions in cases in which there is diagnostic uncertainly clinically.
Beckwith–Wiedemann syndrome is due to epigenetic defects of 11p15 and is characterised by overgrowth, abdominal wall defects and macroglossia. Additional features include ear lobe creases and helical pits, visceromegaly, hemihypertrophy, neonatal hypoglycaemia, renal abnormalities and embryonal tumours (Table 3). Most cases should be straightforward to clinically differentiate from Sotos syndrome as macroglossia is very common in Beckwith–Wiedemann syndrome but has not been reported in Sotos syndrome, whereas characteristic facial features and learning disability are almost universal in Sotos syndrome, but are rare in Beckwith–Wiedemann syndrome. For rare cases that are clinically consistent with both conditions (eg the cases reported by Baujat et al18) molecular analyses at 11p15 and NSD1 should distinguish between the conditions. Individuals with such overlapping phenotypes should be managed according to the underlying molecular defect, that is, individuals with 11p15 imprinting defects should be managed as for Beckwith–Wiedemann syndrome and individuals with NSD1 abnormalities should be managed as for Sotos syndrome.
Benign familial macrocephaly is defined as macrocephaly in an individual with a positive family history of macrocephaly and no neurological deficit.19 No other clinical features are consistently associated with the condition. Benign familial macrocephaly is likely to represent a heterogeneous group of conditions and the diagnosis is reserved for individuals in whom other conditions have been clinically and/or molecularly excluded.
Diagnostic approaches
Before the identification of NSD1, it was recommended that the clinical diagnosis of Sotos syndrome could be supported by bone age assessment and/or cranial imaging.2, 11 However, the bone age is normal or delayed in ∼20% of cases of Sotos syndrome and is a nonspecific test, as advanced bone also occurs in many other conditions.2, 10, 12 Cranial neuro-imaging is abnormal in ∼80% of Sotos syndrome cases but again the observed abnormalities are nonspecific.11 By contrast molecular NSD1 testing provides a simple, safe, sensitive and specific method of confirming a clinical diagnosis of Sotos syndrome in the great majority of cases.10 Molecular testing may not be necessary in some classic Sotos syndrome cases where the clinical diagnosis is confidently made by a clinician experienced in the condition. However, in most cases we recommend that the first-line investigation in a child suspected of having Sotos syndrome should be NSD1 gene testing (Figure 1) and that all individuals in whom an NSD1 abnormality is identified are managed as outlined below. Individuals who meet all three cardinal criteria but do not have an NSD1 abnormality should also be managed as Sotos syndrome. However, we strongly recommend that such cases be reviewed by a clinician with expertise in overgrowth conditions before a diagnosis of Sotos syndrome is given to an individual with a normal NSD1 test result.
Molecular and genetic basis of the disease
NSD1
NSD1, Nuclear receptor SET domain containing protein-1, contains multiple functional domains including two distinct nuclear receptor interaction domains (NID+L and NID−L), five zinc-finger plant homeodomains (PHDI–V), two proline–tryptophan–tryptophan–proline domains, a SET (Su(var)3-9, Enhancer of Zeste and Trithorax) domain, its neighbouring SET-associated cysteine-rich (SAC) domain and a C5HCH motif (Figure 3).20 Relatively little is known of the functions of NSD1 or why its abrogation results in the Sotos phenotype. However, it is likely to play a role in transcriptional regulation through the specific methylation of histone lysine residues (H3-K36 and H4-K20, mediated by SET and SAC domains), the differential binding of the two nuclear receptor interacting domains (NID+L and NID−L) and chromatin–chromatin interactions mediated by the PHD and C5HCH domains.21, 22, 23

Schematic representation of NSD1.
NSD1 mutational mechanisms
Various mutational mechanisms abrogate NSD1 function including truncating mutations, missense mutations, splice-site mutations, partial gene deletions and 5q35 microdeletions.3, 4, 5, 6, 7, 8, 9, 10 The truncating mutations result from small nucleotide insertions and/or deletions or splice-site mutations that result in translational frameshifts and premature stop codons or from base substitutions that generate stop codons (nonsense mutations). The truncating mutations occur throughout the gene. Disease-causing missense mutations are caused by base substitutions within NSD1 functional domains. As these functional domains are primarily located at the 3′ end of NSD1, missense mutations are clustered towards the 3′ end of the gene but there are no mutational hotspots.10 Partial gene deletions account for ∼5% of NSD1 abnormalities and most commonly involve deletion of exons 1 and 2, probably because of the high density of Alu repeats flanking these exons. Some partial gene deletions are generated through nonallelic homologous recombination between flanking Alu repeats whereas others are more likely generated through nonhomologous end joining.24
The mechanism of generation and size of 5q35 microdeletions differ depending upon the ethnic origin of the affected individual. Outside Japan, 5q35 microdeletions are variable in size and predominantly arise through interchromosomal rearrangements.25 In contrast, a uniform 1.9 Mb microdeletion, arising through intrachromosomal rearrangements, has been identified in the majority of Sotos syndrome cases of Japanese descent.26, 27 It is likely that the 1.9 Mb deletion identified in both Japanese and non-Japanese Sotos syndrome cases results from nonallelic homologous recombination between low copy repeat elements flanking NSD1.25, 26, 27, 28 An inversion polymorphism that predisposes to the microdeletion is common in Japan and may explain the high 5q35 microdeletion frequency in the Japanese, although the frequency of the inversion polymorphism outside Japan is currently not known.27 A significant proportion of non-Japanese 5q35 microdeletions do not have breakpoints within known flanking elements and so are likely to have been generated through other mechanisms.25 There is a marked bias towards deletion of the paternally derived allele in all 5q35 microdeletion cases, irrespective of ethnic origin.25, 26 This is likely to reflect, at least in part, the telomeric position of NSD1 as the recombination rate in males is much greater at the 5q telomere compared with that of females.
Contribution of NSD1 to Sotos syndrome
NSD1 abnormalities are identifiable in at least 90% of Sotos syndrome cases.3, 4, 5, 6, 7, 8, 9, 10 In our experience, NSD1 abnormalities do not occur in other human overgrowth conditions, although there are occasional individuals with NSD1 defects that show clinical overlap with Sotos syndrome and other conditions, such as Weaver syndrome and Beckwith–Wiedemann syndrome.4, 10, 18 Among European and American Sotos syndrome cases, intragenic mutations cause 80–85% of Sotos syndrome and 5q35 microdeletions are responsible for only 10–15% of cases.4, 5, 6, 7, 8, 9, 10 In contrast, 5q35 microdeletions are the major mutational mechanism among Japanese Sotos syndrome cases identifiable in >50% of affected individuals.26, 27
Contribution of other genes to Sotos syndrome
NSD1 abnormalities have not been identified in ∼10% of classic Sotos syndrome cases.10 The phenotype of these cases does not differ from Sotos syndrome cases with NSD1 abnormalities and it is likely that they are primarily caused by covert NSD1 mutations that are not detected with current screening techniques. Rare individuals with a clinical diagnosis of Sotos syndrome or Sotos-like syndrome and defects of 11p15 or GPC3 have been described and other molecular defects should be considered in Sotos syndrome cases without NSD1 abnormalities.18, 29 No classic Sotos syndrome case with an abnormality in another gene has been reported.
Genotype–phenotype associations
There is no correlation between mutation position and clinical phenotype and the very broad clinical variability of Sotos syndrome appears independent of genotype as cases with identical mutations often have different clinical features.10 5q35 microdeletion cases are significantly more likely to have severe learning disability than cases with intragenic mutations and also tend to have less pronounced overgrowth.10 This is likely due to the generic effects of microchromosomal defects that are typically associated with learning disability and short stature throughout the genome.30 There is no evidence that deletion of neighbouring genes in 5q35 microdeletion cases has any specific effect on phenotype and all features observed in microdeletion cases have also been reported in individuals with intragenic NSD1 mutations.10, 25
Inheritance of NSD1 abnormalities
The great majority of individuals with Sotos syndrome do not have a similarly affected relative. In addition, extensive NSD1 mutation analyses of unaffected parents demonstrated that none carried the mutation present in their child, indicating that Sotos syndrome is a fully penetrant, primarily sporadic disorder.10
Rare familial Sotos syndrome pedigrees (<10%) with NSD1 mutations have been reported.10, 26, 31 Such families often harbour missense mutations, although families with truncating mutations are known. The phenotype within families can be variable. The paucity of familial Sotos pedigrees indicates that the vertical transmission rate of NSD1 defects is very low. The reasons for this are not entirely clear.
Management
Genetic counselling
Most individuals with Sotos syndrome are the result of de novo mutations. No affected siblings of unaffected parents have been reported indicating that the incidence of germline mosaicism must be low. Therefore, the recurrence risk of unaffected parents approximates to the population risk, which is estimated to be ∼1:15 000.
Affected individuals should be counselled with an offspring risk of 50% as for other autosomal dominant conditions. The observed vertical transmission rate in Sotos syndrome is clearly lower than this as familial Sotos syndrome is rare, with less than 10% of cases having an affected relative. However, in our limited experience of adult Sotos syndrome cases that have had children, approximately 50% of the offspring have been affected and there is currently no obvious evidence of impaired fertility, increased fetal loss or increased morbidity/mortality in adulthood.
Initial assessment of individuals with Sotos syndrome
At the initial assessment of the individual with an NSD1 abnormality or a high clinical suspicion of Sotos syndrome, the history and examination should aim to identify known associations/complications of the condition (Figure 4). Of particular note, we recommend that the clinician perform investigations to identify renal or cardiac anomalies that, if undetected, may lead to significant morbidity. Baseline renal investigations should include dipstick urinalysis, blood pressure measurement and renal ultrasound scan, and we also recommend a baseline cardiac echocardiogram together with cardiac auscultation and blood pressure measurement. If abnormalities are detected on these routine investigations referral to the appropriate specialist should be considered.

Suggested approach to the initial evaluation and management of the individuals with Sotos syndrome.
Surveillance of individuals with Sotos syndrome
The primary role of the clinical geneticist will be to make the diagnosis and perform the initial evaluation and to discuss recurrence and offspring risks. The clinical geneticist may also be valuable in coordination of the various specialities involved in the care of individuals with complex medical problems.
In general, individuals with Sotos syndrome would benefit from annual review. This review could be performed by the family doctor or general paediatrician depending on the age of the individual and how severely affected they are. The review should involve a thorough history, examination including cardiac auscultation, blood pressure measurement, back examination and a dipstick urinalysis. If abnormalities are detected, the appropriate specialist referral(s) should be made.
Tumour screening is not recommended in individuals with Sotos syndrome. Very few individuals with Sotos syndrome develop tumours.10 The relative risk of neural crest tumours, saccrococcygeal teratomas and possibly some haematological malignancies is increased, but the absolute risk of tumour development in Sotos syndrome is <3%.10 Moreover, the spectrum of tumour types reported is broad and does not include tumours for which there are validated screening protocols. Of note, extensive review of hundreds of (molecularly confirmed) Sotos syndrome cases demonstrates that Wilms tumour occurs very rarely and thus Wilms tumour screening is not warranted.
Conclusion
Since haploinsufficiency of NSD1 was shown to cause Sotos syndrome in 2002, the NSD1 mutational spectrum has been defined, the phenotype of Sotos syndrome clarified and diagnostic and management guidelines developed. However, several questions remain unresolved. The normal functions of NSD1 are largely unknown and how functional NSD1 abrogation results in the diverse clinical features of Sotos syndrome is unclear.
From a clinical perspective, the low vertical transmission rate is unexplained and of considerable importance to the many individuals with Sotos syndrome and mild learning disability who may consider having children in the future. Long-term prospective follow-up of affected individuals will likely be required to elucidate this. Such studies will also be invaluable in clarifying the phenotype and long-term outcome of Sotos syndrome in adults for which limited information is currently available.
References
Sotos JF, Dodge PR, Muirhead DD, Crawford JD, Talbot NB : A syndrome of excessively rapid growth and acromegalic features and a nonprogressive neurologic disorder. N Engl J Med 1964; 271: 109–116.
Cole TR, Hughes H : Sotos syndrome: a study of the diagnostic criteria and natural history. J Med Genet 1994; 31: 20–32.
Kurotaki N, Imaizumi K, Harada N et al: Haploinsufficiency of NSD1 causes Sotos syndrome. Nat Genet 2002; 30: 365–366.
Douglas J, Hanks S, Temple IK et al: NSD1 mutations are the major cause of Sotos syndrome and occur in some cases of Weaver syndrome but are rare in other overgrowth phenotypes. Am J Hum Genet 2003; 72: 132–143.
Rio M, Clech L, Amiel J et al: Spectrum of NSD1 mutations in Sotos and Weaver syndromes. J Med Genet 2003; 40: 436–440.
Turkmen S, Gillessen-Kaesbach G, Meinecke P et al: Mutations in NSD1 are responsible for Sotos syndrome, but are not a frequent finding in other overgrowth phenotypes. Eur J Hum Genet 2003; 11: 858–865.
Cecconi M, Forzano F, Milani D et al: Mutation analysis of the NSD1 gene in a group of 59 patients with congenital overgrowth. Am J Med Genet A 2005; 134: 247–253.
Waggoner DJ, Raca G, Welch K et al: NSD1 analysis for Sotos syndrome – insights and perspectives from the clinical laboratory. Genet Med 2006; 8: 379–382.
Melchior L, Schwartz M, Duno M : dHPLC screening of the NSD1 gene identifies nine novel mutations-summary of the first 100 Sotos syndrome mutations. Ann Hum Genet 2005; 69: 222–226.
Tatton-Brown K, Douglas J, Coleman K et al: Genotype–phenotype associations in Sotossyndrome: an analysis of 266 individuals with NSD1 aberrations. Am J Hum Genet 2005; 77: 193–204.
Schaefer GB, Bodensteiner JB, Buehler BA, Lin A, Cole TR : The neuroimaging findings in Sotos syndrome. Am J Med Genet 1997; 68: 462–465.
Tatton-Brown K, Rahman N : Clinical features of NSD1-positive Sotos syndrome. Clin Dysmorphol 2004; 13: 199–204.
Weaver DD, Graham CB, Thomas IT, Smith DW : A new overgrowth syndrome with accelerated skeletal maturation, unusual facies, and camptodactyly. J Pediatr 1974; 84: 547–552.
Marsh DJ, Kum JB, Lunetta KL et al: PTEN mutation spectrum and genotype-phenotype correlations in Bannayan–Riley–Ruvalcaba syndrome suggest a single entity with Cowden syndrome. Hum Mol Genet 1999; 8: 1461–1472.
Bannayan GA : Lipomatosis, angiomatosis, and macrencephalia. A previously undescribed congenital syndrome. Arch Pathol 1971; 92: 1–5.
Riley Jr HD, Smith WR : Macrocephaly, pseudopapilledema and multiple hemangiomata: a previously undescribed familial syndrome. Pediatrics 1960; 26: 293–300.
Ruvalcaba RH, Myhre S, Smith DW : Sotos syndrome with intestinal polyposis and pigmentary changes of the genitalia. Clin Genet 1980; 18: 413–416.
Baujat G, Rio M, Rossignol S et al: Paradoxical NSD1 mutations in Beckwith–Wiedemann syndrome and 11p15 anomalies in Sotos syndrome. Am J Hum Genet 2004; 74: 715–720.
Cole TR, Hughes HE : Autosomal dominant macrocephaly: benign familial macrocephaly or a new syndrome? Am J Med Genet 1991; 41: 115–124.
Kurotaki N, Harada N, Yoshiura K, Sugano S, Niikawa N, Matsumoto N : Molecular characterization of NSD1, a human homologue of the mouse Nsd1 gene. Gene 2001; 279: 197–204.
Rayasam GV, Wendling O, Angrand PO et al: NSD1 is essential for early post-implantation development and has a catalytically active SET domain. EMBO J 2003; 22: 3153–3163.
Huang N, vom BE, Garnier JM et al: Two distinct nuclear receptor interaction domains in NSD1, a novel SET protein that exhibits characteristics of both corepressors and coactivators. EMBO J 1998; 17: 3398–3412.
Stec I, Nagl SB, van Ommen GJ, den Dunnen JT : The PWWP domain: a potential protein-protein interaction domain in nuclear proteins influencing differentiation? FEBS Lett 2000; 473: 1–5.
Douglas J, Tatton-Brown K, Coleman K et al: Partial NSD1 deletions cause 5% of Sotos syndrome and are readily identifiable by multiplex ligation dependent probe amplification. J Med Genet 2005; 42: e56.
Tatton-Brown K, Douglas J, Coleman K et al: Multiple mechanisms are implicated in the generation of 5q35 microdeletions in Sotos Syndrome. J Med Genet 2005; 42: 307–313.
Kurotaki N, Harada N, Shimokawa O et al: Fifty microdeletions among 112 cases of Sotos syndrome: Low copy repeats possibly mediate the common deletion. Hum Mutat 2003; 22: 378–387.
Visser R, Shimokawa O, Harada N et al: Identification of a 3.0-kb major recombination hotspot in patients with Sotos syndrome who carry a common 1.9-Mb microdeletion. Am J Hum Genet 2005; 76: 52–67.
Lupski JR : Genomic disorders: structural features of the genome can lead to DNA rearrangements and human disease traits. Trends Genet 1998; 14: 417–422.
Li M, Shuman C, Fei YL et al: GPC3 mutation analysis in a spectrum of patients with overgrowth expands the phenotype of Simpson–Golabi–Behmel syndrome. Am J Med Genet 2001; 102: 161–168.
Devriendt K, Vermeesch JR : Chromosomal phenotypes and submicroscopic abnormalities. Hum Genomics 2004; 1: 126–133.
Hoglund P, Kurotaki N, Kytola S, Miyake N, Somer M, Matsumoto N : Familial Sotos syndrome is caused by a novel 1 bp deletion of the NSD1 gene. J Med Genet 2003; 40: 51–54.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Tatton-Brown, K., Rahman, N. Sotos syndrome. Eur J Hum Genet 15, 264–271 (2007). https://doi.org/10.1038/sj.ejhg.5201686
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.ejhg.5201686
Keywords
This article is cited by
-
Tall stature and gigantism in transition age: clinical and genetic aspects—a literature review and recommendations
Journal of Endocrinological Investigation (2023)
-
Cerebrovascular diseases in two patients with entire NSD1 deletion
Human Genome Variation (2021)
-
Drosophila NSD deletion induces developmental anomalies similar to those seen in Sotos syndrome 1 patients
Genes & Genomics (2021)
-
First report of tethered cord syndrome in a patient with Sotos syndrome
BMC Pediatrics (2020)
-
Berlin Heart EXCOR® pediatric ventricular assist device in a patient with Sotos syndrome: a case report
Journal of Medical Case Reports (2019)
