
Abstract
A novel transmitted 2–3 Mb deletion of 2q14.1–q14.2 was found in an affected boy from a consanguineous family with a possible diagnosis of PEHO syndrome (OMIM 260565). BAC FISH showed that the deletion included a minimum of 20 genes including the homeobox engrailed 1 gene (EN1). However, the same deletion was also found in his phenotypically normal father and brother (family 1). The phenotype of the proband may, therefore, have been coincidental to the deletion, a result of a recessive condition within or outside the deleted segment or possibly due to variable dosage compensation of EN1 by the paralogous EN2 gene at 7q36. BAC FISH also showed that this deletion overlapped with a previously reported transmitted deletion of 2q13–q14.1 that had no phenotypic consequences (family 2). The deleted regions contained a total of 32 genes and comprise the final 5.25 Mb of the ancestral chromosome 2B from which chromosome 2 was formed in man. These families provide further evidence that heterozygous deletions of regions of low gene density are compatible with a normal phenotype.
Similar content being viewed by others


Introduction
A growing number of relatively small imbalances are being detected using high resolution conventional cytogenetics, high-resolution comparative genomic hybridisation (HR-CGH)1 and array CGH.2, 3, 4, 5 Over 10% of the abnormalities detected using HR-CGH1 and over 40% of those detected using array CGH2, 3, 4, 5 are directly transmitted from parents to children. In a recent review, 130 families with directly transmitted unbalanced chromosome abnormalities were identified6 and, in the majority of these (77/130), both probands and parents were phenotypically affected. However, in 30/130, an unaffected parent had the same imbalance as an affected child and, in 23/130, both the probands and parents were phenotypically normal. Here, we present further examples of an affected boy with the same 2–3 Mb deletion of 2q14.1–q14.2 as his unaffected father and brother. This deletion overlapped with a previous transmitted deletion of 2q13–q14.1 in a phenotypically normal mother and daughter7 which has now been mapped in more detail.
Methods
G-banded chromosomes were analysed at the 550 or higher band level and FISH was carried out with 1 Megabase (Mb) clones or tiling path BACs selected from the Ensembl database (www.ensembl.org/Homo_sapiens/cytoview). Karyotype results include the probes informative for the breakpoints only. Genes assigned to the deleted regions were obtained from the Ensembl database (v32) using the MartView data export tool (www.ensembl.org/Multi/martview). Messenger RNA homologies were obtained using the NCBI bl2seq alignment tool (www.ncbi.nlm.nih.gov/blast/bl2seq/wblast2.cgiBLASTN).
Family report and results
Family 1 del(2)(q14.1q14.2)
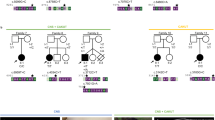
The proband was the sixth child born to consanguineous Pakistani parents. His mother had no history of miscarriages. The proband presented at birth with a left congenitally dislocated hip and minor dysmorphic features including long nails, overlapping fingers and toes and an unusual right ear with an overfolded helix and double ear lobe creases. At the age of 14 months, he had profound developmental delay, seizures, head circumference just below the 0.4th centile, marked hypertonia, puffy hands and feet and roving eye movements with hypoplastic optic discs. MRI scan of his brain showed simplified gyral formation and a slightly small cerebellum. A diagnosis of progressive encephalopathy with oedema, hypsarrythmia and optic atrophy (PEHO) syndrome (OMIM 260565) has been suggested.
One sister had died previously in infancy with a history of Dandy Walker malformation, profound developmental delay, brisk reflexes, roving eye movements and bilateral congenital dislocation of the hips. Although the CT scan findings were reportedly different to those of the proband, it is possible that both affected children had the same condition.
A deletion of 2q14.1 to 2q14.2 was found in the affected proband as well as his phenotypically normal father and 16-year-old brother (Figure 1). The brother attends mainstream school and is doing well academically. The father drives a minibus and has no history of learning difficulties at school. Two further brothers (aged 8 and 18) had normal chromosomes. The karyotype of the affected sister was reported as normal at the 400–550 band level of resolution but this may not have been sufficient to detect the subtle deletion of 2q and the slides were no longer available for retrospective review.

Partial karyotype of the deletion in family 1 with the deleted region indicated by black arrows. Note the reduction in the intensity of band 2q14.1 and the reduction in size of band 2q14.2 on the right hand chromosome 2 (and the magnified image of the same chromosome).
Using FISH on the proband, three clones from 2q14.1 and 2q14.2 were deleted (Figure 2) and all other probes gave normal results (Table 1). The proximal breakpoint was within the ∼0.7 Mb region between 252I18 and 59P4 and the distal breakpoint within or close to the 2 kb overlapping region of 19E11 and 77A13. The karyotype was therefore 46,XY,del(2)(q14.1q14.2).ish del(2)(q14.1q14.2)(252I18+,59P4−,19E11−,77A13+). FISH with probes from opposite ends of the deletion confirmed that the deletion in his father and brother were of the same extent. The deletions had a minimum size of 2.24 Mb and a maximum size of 2.91 Mb. The minimal deleted region contains five known and 15 novel genes (Table 2) including the EN1 homeobox engrailed 1 gene, which is 119.3 Mb from the telomere and maps to the proximal end of the deleted BAC 19E11 (119.3–119.4 Mb).

(a–d) Dual colour BAC FISH images of the deletions in families 1 and 2; normal red (TRITC) signals with (a) 395L14 and the absent signals with (b) 432G15 from the mother of family 1; normal signals with (c) 19E11 and absent signals with (d) 59P4 from the father of family 2. White arrows indicate the position of the missing signals and the green (FITC) signals are from the chromosome 2 centromere probe pBS4D in each case.
Family 2 del(2)(q13q14.1)
This deletion of 2q13 to 2q14.1 was originally ascertained in a female of 38 after three miscarriages. She had two children already and her pregnancy at the time was chromosomally normal. The same deletion was found in her mother who had two children and no history of miscarriages.7 YACs 786a12 and 817b4 were deleted, 791f4 partially deleted and the deletion estimated at 4.5–6.0 Mb. In order to test the hypothesis that this deletion was the same as that in family 1, we remapped the deletion and found seven clones were deleted (Table 1). The proximal breakpoint was mapped to the 22 kb region of overlap between BACs 395L14 and 432G15 in band 2q14.1 in both mother and daughter. This breakpoint is adjacent to the ancestral great ape chromosome fusion site (2qFus) which is contained within BAC 395L14 close to the overlapping segment of 432G15.8 The distal breakpoint was within the 1 Mb between BACs 59P4 and 98C1 within the same band. The karyotype was, therefore, revised to 46,XX,del(2)(q13q14.1).ish del(2)(q14.1q14.1)(395L14+,432G15−,59P4−,98C1+). The minimum size of the deletion was 3.21 Mb and the maximum size 4.27 Mb. The minimum interval contains four known and eight novel genes (Table 2).
The minimum combined length of the deletions in families 1 and 2 was ∼5.25 Mb containing 32 genes of which nine were known and 23 novel (Table 2). The maximum size of the overlap between the two deletions was a gene poor region of ∼1.9 Mb.
Discussion
The combined results from the overlapping 2q deletions in families 1 and 2 imply that 5.25 Mb from 2q13 to 2q14.2 including 32 genes can be deleted without phenotypic consequences. However, in family 1, the same 2–3 Mb deletion was present in the affected proband and his phenotypically normal father and sibling. There are at least three possible explanations for this apparent discrepancy:
-
1
The deletion is coincidental to the phenotype of the proband (and possibly his sister) and the suggested diagnosis of PEHO results from an independent recessive condition in this consanguineous family.
-
2
The deletion may have unmasked a recessive allele at one of the 20 genes within the deleted segment despite the fact that it represents only ∼0.1% of the genome.
-
3
A single copy of the homeobox engrailed 1 (EN1) transcription factor (OMIM 131290) gene may be more susceptible to variations in the timing or level of its expression or to variable dosage compensation by its EN2 paralog in 7q36 (OMIM 131310).
EN1 is an attractive candidate gene as En1 behaves in a dosage sensitive manner in the mouse, is directly involved in cerebellar phenotypes9 (the cerebellum was slightly smaller in the proband), has a role in the direction of foliation9 (gyral formation was simplified in the proband) and combines with En2 in the formation of the optic tectum10 (the proband had hypoplastic optic discs). Evidence both for11 and against12 an association between autistic spectrum disorder and SNPs at the paralogous EN2 gene has also been presented but it is not clear that upregulation of EN2 could account for the developmental delay found in the proband.
Nevertheless, family 1 indicates that heterozygous deletions that include EN1 are compatible with an apparently normal phenotype in man. Several of the other known genes within the deleted regions in families 1 and 2 also have related genes elsewhere (Table 2). It is therefore possible that hemizygosity for these genes is more easily tolerated as suggested for haplosufficient deletions of 2p12.13 In this context, it is interesting that replacement of En1 with En2 sequences rescues the En-1 mutant phenotype in the mouse14 and that dosage compensation of En1 by En2 has already been invoked to explain the suppression of the En-1 cerebellar phenotype by a change of genetic background.9 Human EN1 (NM_001426) and EN2 (NM_001427) are 82% homologous at the mRNA level and, although expression of EN2 is more restricted than that of EN1, upregulation of paralogous loci has been recorded in single gene knockouts in yeast, even when the paralogs are not coexpressed.15 The insulin induced protein 2 (INSIG2) gene (NM_016133) is also between 75 and 83% homologous with the three transcript variants of the INSIG1 paralog (NM_198337, 198336 and 005542) which maps with EN2 to 7q36.
The proband in family 2 had been ascertained for miscarriages,7 but it is difficult to see how these could have been caused by a deletion that is compatible with a normal phenotype, especially when the carrier mother had no miscarriages herself and most simple deletions are transmitted without causing additional imbalances.6
These deletions are immediately adjacent to the site at which the ancestral great ape chromosomes 2A and 2B fused to produce human chromosome 2.8 Thus, the terminal orthologous 5.25 Mb of the short arm of the ancestral chromosome 2B (PTR 13 in chimpanzee) can apparently be deleted in humans and gain or loss of 32 subtelomeric genes does not necessarily have detectable phenotypic consequences.16 The 2q fusion region is believed to be inherently unstable 17 but, without precise identification of the breakpoint, we can only speculate that this might have contributed to the formation of the proximal breakpoint in family 2.
In conclusion, family 1 illustrates the difficulties in making a clear diagnosis even when the gene content of a particular imbalance is known. Nevertheless, families 1 and 2 provide further evidence that deletions of regions of low gene density are compatible with a normal phenotype.13 These results will be added to the Chromosome Anomaly Collection of transmitted imbalances for future reference (www.ngrl.org.uk/Wessex/collection/).
References
Kirchhoff M, Pedersen S, Kjeldsen E et al: Prospective study comparing HR-CGH and subtelomeric FISH for investigation of individuals with mental retardation and dysmorphic features and an update of a study using only HR-CGH. Am J Med Genet 2004; 127A: 111–117.
Vissers LE, de Vries BB, Osoegawa K et al: Array-based comparative genomic hybridization for the genomewide detection of submicroscopic chromosomal abnormalities. Am J Hum Genet 2003; 73: 1261–1270.
Shaw-Smith C, Redon R, Rickman L et al: Microarray based comparative genomic hybridisation (array-CGH) detects submicroscopic chromosomal deletions and duplications in patients with learning disability/mental retardation and dysmorphic features. J Med Genet 2004; 41: 241–248.
Schoumans J, Ruivenkamp C, Holmberg E, Kyllerman M, Anderlid BM, Nordenskjold M : Detection of chromosomal imbalances in children with idiopathic mental retardation by array based comparative genomic hybridisation (array-CGH). J Med Genet 2005; 42: 699–705.
Rosenberg C, Knijnenburg J, Bakker E et al: Array-CGH detection of micro rearrangements in mentally retarded individuals: clinical significance of imbalances present both in affected children and normal parents. J Med Genet 2005, Jun 24:[E-pub ahead of print].
Barber JCK : Directly transmitted unbalanced chromosome abnormalities and euchromatic variants. J Med Genet 2005; 42: 609–629.
Sumption ND, Barber JCK : Transmitted deletion of 2q13 to 2q14.1 causes no phenotypic abnormalities. J Med Genet 2001; 38: 125–126.
Fan Y, Linardopoulou E, Friedman C, Williams E, Trask BJ : Genomic structure and evolution of the ancestral chromosome fusion site in 2q13–2q14.1 and paralogous regions on other human chromosomes. Genome Res 2002; 12: 1651–1662.
Bilovocky NA, Romito-DiGiacomo RR, Murcia CL, Maricich SM, Herrup K : Factors in the genetic background suppress the engrailed-1 cerebellar phenotype. J Neurosci 2003; 23: 5105–5112.
Nakamura H : Regionalisation and acquisition of polarity in the optic tectum. Prog Neurobiol 2001; 65: 473–488.
Benayed R, Gharani N, Rossman I et al: Support for the Homeobox Transcription Factor Gene ENGRAILED 2 as an Autism Spectrum Disorder Susceptibility Locus. Am J Hum Genet 2005; 77: 851–868.
Zhong HH, Serajee FJ, Nabi R, Mahbubul Huq AHM : No association between the EN2 gene and autistic disorder. J Med Genet 2003; 40: e4.
Barber JCK, Thomas NS, Collinson MN et al: Segmental haplosufficiency; transmitted deletions of 2p12 include a pancreatic regeneration gene cluster and have no apparent phenotypic consequences. Eur J Hum Genet 2005; 13: 278–282.
Hanks M, Wurst W, Anson-Cartwright L, Auerbach AB, Joyner AL : Rescue of the En-1 mutant phenotype by replacement of En-1 with En-2. Science 1995; 269: 679–682.
Kafri R, Bar-Even A, Pilpel Y : Transcription control reprogramming in genetic backup circuits. Nat Genet 2005; 37: 295–299.
Linardopoulou EV, Williams EM, Fan Y, Friedman C, Young JM, Trask BJ : Human subtelomeres are hot spots of interchromosomal recombination and segmental duplication. Nature 2005; 437: 94–100.
Cheng Z, Ventura M, She X et al: A genome-wide comparison of recent chimpanzee and human segmental duplications. Nature 2005; 437: 88–93.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Barber, J., Maloney, V., Bewes, B. et al. Deletions of 2q14 that include the homeobox engrailed 1 (EN1) transcription factor are compatible with a normal phenotype. Eur J Hum Genet 14, 739–743 (2006). https://doi.org/10.1038/sj.ejhg.5201605
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.ejhg.5201605
Keywords
This article is cited by
-
A de novo 10.79 Mb interstitial deletion at 2q13q14.2 involving PAX8 causing hypothyroidism and mullerian agenesis: a novel case report and literature review
Molecular Cytogenetics (2014)
-
Characterization of two ectrodactyly-associated translocation breakpoints separated by 2.5 Mb on chromosome 2q14.1–q14.2
European Journal of Human Genetics (2009)
