
Abstract
Asthma is a complex disease and the intricate interplay between genetic and environmental factors underlies the overall phenotype of the disease. Families with at least two siblings with asthma were collected from Europe, Australia and the US. A genome scan using a set of 364 families with a panel of 396 microsatellite markers was conducted. Nonparametric linkage analyses were conducted for asthma and three asthma-related phenotypes: bronchial hyper-reactivity (BHR), strict definition of asthma and atopic asthma. Nine chromosomal regions with LOD scores greater than 1.5 were identified (chromosomes 1q, 2p, 3q, 4p, 4q, 6q, 12q, 20p and 21). Linkage refinement analysis was performed for three BHR loci by genotyping single nucleotide polymorphisms at an average marker density of 1 cM. The LOD scores increased to 3.07 at chromosome 4p and 4.58 at chromosome 2p, while the chromosome 6p locus did not refine. The LOD score at the chromosome 2p locus is highly significant on a genome-wide basis. The refined locus covers a region with a physical size of 12.2 Mb. Taken together, these results provide evidence for a major asthma susceptibility locus on chromosome 2p.
Similar content being viewed by others


Introduction
In the UK, the prevalence of diagnosed asthma and symptoms strongly suggestive of asthma in children has increased at a rate of about 5% a year and this trend was noticed in most of the countries studied.1 The cost of treating asthma is approximately $6 billion per annum in the US.2, 3 The complex interplay between genetic and environmental factors underlies the overall phenotype of asthma. Environmental exposure to allergens, pollutants, and respiratory viruses contribute to the development of asthma.4, 5 Genome-wide screens for asthma and related phenotypes have been completed in a number of independent populations.6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16 The regions most consistently shown to be in linkage with asthma include the cytokine cluster on chromosome 5 and chromosome 11q13, 12q24, 16q23, and 17q12 (reviewed in Cookson17 and Wills-Karp and Ewart18). Recently, there have been four reports on the positional cloning of asthma genes.19, 20, 21, 22 Overall results from these genome scans differ significantly, which may be attributed to the differences in sample size and the choice of phenotypes. Replication of linkage findings in complex diseases requires a substantially larger data set than that in which the original findings were made; this is because practicable sample sizes always represent a fraction of the ideal, and some chance concentration of families with linkage in a particular region is likely.23 Moreover, subtle differences in the phenotype definition between studies render the replication of linkage results difficult.
Herein, we present results of a genome-wide search for linkage to asthma from a large, international family-based study, using a set of well-validated markers. In comparison to previously reported asthma linkage results, this study is larger in sample size and is based on a wide array of demographic and clinical variables. Moreover, the use of multiple asthma case definitions enabled us to compare identified linkage regions across these phenotypic subgroups, as well as interpreting our results in light of previously published reports.
Materials and methods
Subject recruitment and clinical evaluation
Asthma families were studied at eight clinical centers (see online supplementary Table 1 for details) as part of the Genetics of Asthma International Network (GAIN) collection.24 The following criteria were used for the ascertainment of the families:
-
a)
Male and female probands aged 8–35 years, with documented episodes of wheezing in the past 12 months and who were physician diagnosed as having asthma.
-
b)
Affected sibling/s aged 7–35 years who had documented symptoms and treatment for asthma for at least 2 years.
The asthma diagnosis was defined by the participating physician who recruited the patient and not from self-reports. Baseline spirometry was performed according to American Thoracic Society criteria.25, 26 A methacholine challenge test was carried out on all subjects unless the baseline FEV1 was ≤70% of the predicted value.27, 28 A bronchodilator reversibility test was performed in those subjects with baseline FEV1 ≤70% of the predicted value. Skin prick tests for common allergens (dust mites – Dermatophagoides pteronyssinus and D. farinae, dog, cat, grass mix, pollen and alternaria) were conducted. Allergens were procured from ALK, Reading, Berks, UK. The patients were considered atopic if the wheal diameter was ≥3 mm above the negative control (saline) for at least one allergen tested. Total serum IgE measurements were also taken. The clinical, laboratory and questionnaire data and the DNA samples were collected from the siblings and the parents. Appropriate Institutional Review Boards approved the studies and informed consent was obtained from all subjects. All of the families used in the linkage analysis from this population were of Caucasian ethnicity.
Quality control of the asthma diagnosis
Families were ascertained from different clinical centers in the world and physicians trained in several continents assigned the diagnosis. Before conducting genetic analyses, the asthma phenotype definitions were validated in order to minimize Type I and Type II errors. A clinical panel comprised of seven physicians was established and clinical data from 200 individuals from the participating sites were provided to these physicians. The panel was asked to assign the patients into the following diagnostic categories only by looking at the data provided. (1) Definite asthma, (2) probable asthma (very likely that the patient has asthma but the available data leaves some doubt), (3) possible asthma (likely not to have asthma but there is some data that could support the diagnosis) and (4) no asthma.
Phenotypes
The following phenotypes were evaluated for linkage analysis. (a) Asthma (diagnosis by a participating physician); (b) bronchial hyper-reactivity (BHR, a positive methacholine response (≥20% reduction in FEV1) at or below 8 mg/ml of methacholine); (c) atopic asthma (asthma and a positive result on at least one skin allergen test); (d) strict asthma (two or more classic symptoms – cough, wheeze and dyspnea – and a positive methacholine challenge test or bronchodilator reversibility based on the algorithm proposed by the Collaborative Study on the Genetics of Asthma29, 30).The numbers of informative sib pairs for asthma, BHR, strict asthma and atopic asthma were 406, 184, 158 and 334, respectively. Hence, the power of the study to detect linkage for asthma and atopic asthma is substantial but goes down for the other sub phenotypes.
Genotyping
Amplification of microsatellite markers
A genome-wide microsatellite marker scan was performed on genomic DNA extracted from whole blood using the Puregene© system (Gentra systems). The genotyping methodology used for the microsatellite markers is described elsewhere.31 The genotyping was done at the Center for Human Genetics at Duke University, North Carolina. Potential pedigree errors were checked using the program RELPAIR.32 Follow-up analyses of sample integrity were performed with PREST.33
SNP genotyping
For refinement of linkage regions, marker spacing was decreased to approximately 1 cM intervals across loci of interest. Single nucleotide polymorphism (SNP) markers with minor allele frequency of >10% were selected from public databases and from the Applied Biosystems Assays-on-Demand™ catalogue. SNP genotyping was performed using Amplifluor and Taqman® technologies.
Statistical analysis
Inter-rater reliability of asthma diagnosis by the panel of physicians was analyzed using the MAGREE34 macro in SAS software. Inspection of Mendelian errors over all markers and the distribution of marker alleles shared identical by state and identical by descent for relative pairs was used to check for pedigree errors and monozygotic twins.33 Any family member whose biological relationship was found to be in error was excluded from analysis. Mendelian errors were eliminated using an algorithm which identifies individuals whereby setting their genotype for a particular marker to ‘missing’ eliminates the errors (Ramana Idury, personal communication).
All markers typed were placed on the Decode-NCBI integrated map. Multipoint nonparametric linkage analysis, as implemented in MERLIN, was used in all analyses.35 This software implements the Kong and Cox's likelihood modification36 which calculates exact likelihoods and reports LOD scores based on a one parameter allele sharing statistic, Zlr, rather than using the perfect data approximation. LOD scores were computed at a grid density of 1 and 0.1 cM for the primary and refinement scans, respectively. Entropy, a measure of information content, was also calculated in Merlin. As a first pass, an arbitrary and modest LOD threshold was chosen for declaring a linkage region as ‘interesting’. For example, each highlighted region has at least one LOD score >1.5 from any of the four phenotypes analyzed. While this approach may increase the number of false-positive results that may be subjected to more detailed examination, it decreases the serious possibility of missing a true genetic effect. The span of the peaks (in cM) was reported as 1 LOD drop support interval from the highest LOD score in the Decode-NCBI integrated map.
Results
Clinical features
The families for the asthma linkage scan reported here were recruited from eight clinical centers in Europe, Australia and the US. The clinical features of the subjects included in the genome scan are reported in Table 1. The average number of individuals per family was 4.3 (76% families with four individuals, 21% with five individuals and 3% with six individuals). A summary of the phenotype data used in the linkage analysis for siblings and parents are shown in Table 2. Ninety three percent of the siblings included in the linkage scan were diagnosed as asthmatics by the participating physician who recruited the patients. Forty eight (5.9%) siblings had a history of asthma, but the participating physician diagnosed them as non-asthmatics. One percent (n=8) of the siblings did not receive a conclusive diagnosis of asthma. Among the siblings with a physician's diagnosis of asthma, 58% (n=448) were bronchial hyperreactive, while 32% (n=243) were not. Fifty eight percent (n=445) of the siblings with a physician's diagnosis of asthma were positive for the strict definition of asthma and 6% (n=47) were negative, while 36% (n=278) did not have a definitive diagnosis. Seventy three percent of the siblings (n=558) with a physician's diagnosis of asthma also had positive reaction to one or more allergens.
Quality control of asthma diagnosis
Statistical analysis of the data for inter-rater variability showed that there was strong agreement between the panel physicians on the diagnosis. The multiple Kappa statistic for the inter-rater reliability was 0.42 (P-value <0.0001). As expected, there was more agreement between the physicians on the extreme responses (definite asthma and definitely no asthma). The panel diagnosis was compared with the original diagnosis and there was very good agreement between the majority decision in the panel and the diagnosis by the physician who recruited the patients (details of the concordance in diagnosis is available in online supplementary Table 2). Thus, we have established that there is substantial reliability in assignment of the disease status among the participating investigators.
Linkage analysis
A total of 1551 samples were genotyped using a panel of (396) STRP markers. A list of the markers and the map distances are provided in the online supplementary Table 3. DNA samples were not available for four parents with phenotype data. Inspection of the pedigree errors33 indicated that 18 families had at least one error, which translated to an error rate of 0.14%. Corrections were made to the families in error. Only two families were dropped entirely from the analyses.
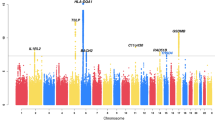
These data were analyzed for linkage to four asthma-related phenotypes using multipoint nonparametric linkage. The results of the multipoint linkage analysis of the combined scan are shown in Figure 1. LOD scores were plotted versus the map distance for each of the 22 autosomes and the X chromosome. Nine chromosomal regions with a LOD score greater than 1.5 for at least one of the four phenotypes were identified (chromosome 1q, 2p, 3q, 4p, 4q, 6q, 12q, 20 and 21, see Table 3 for details).


Genome-wide scan of asthma phenotypes in the Genetics of International Network (GAIN) Families. Multipoint LOD score is shown on the vertical axis and map distance on the horizontal axis. The list of the STRP markers analyzed in the genome scan and the map distances are given in the supplementary Table 3.
Linkage refinement analysis
Three loci showing suggestive evidence of linkage to the BHR phenotype were selected for the follow-up study (2p, 4p and 6q). SNP markers at 1 cM interval were genotyped to cover the entire peak (not just the 1 LOD support interval). The average linkage information content of these regions increased from 80 to 90% after adding the SNP markers. Map distances were obtained from the NCBI Build 33 using the Decode STR map as a framework. These maps were checked for consistency in marker order with other available mapping resources, for example, Marshfield and Genethon Maps. To ensure that observed increase in linkage LOD score for the fine-mapping study are robust and not due to SNPs in LD with each other,37, 38 LD across the region was tested using 724 founders with genotype data. On average, the refinement regions had a marker every cM. LD across the refined regions was low and it is unlikely that the LOD score would have been inflated by the effects of LD.
The multipoint LOD score for BHR increased to 4.58 (P<0.000001; Figure 2) on chromosome 2, after adding 85 SNPs in the region of interest. The LOD scores at this locus are highly significant on a genome-wide basis, even allowing for multiple marker testing. Similarly, 36 SNPs were used to fine map the chromosome 4p region. There was only a modest increase in the LOD score for BHR at this locus on refinement, increasing from 2.75 to 3.07 (Figure 3). Eighty five SNPs were used to perform the linkage refinement analysis of the chromosome 6q locus; however, this locus did not yield any refined peaks (see figure in the online supplementary data for details). The maximum multipoint LOD score for atopic asthma increased from 1.77 to 2.59 at 146 cM. However, the LOD score for BHR decreased from 1.7 to 1.2 and the peak LOD score shifted to 130 cM (original Max LOD was at 126 cM). There were multiple peaks for other phenotypes and the results in general indicate that the 6q locus did not refine upon adding more markers. The linkage information content on chromosome 2p, 4p and 6p are also shown in Figures 2 and 3 and online supplementary figure, respectively. The list of the markers used for the linkage refinement analysis of chromosome 2p, 4p and 6q regions are provided in the online supplementary Tables 4, 5 and 6, respectively.

Linkage refinement analysis of the chromosome 2 BHR locus. Eighty five single nucleotide polymorphism (SNP) markers were genotyped in the GAIN collection and the data was analyzed by multipoint nonparametric linkage methods. LOD scores and information content are shown in the vertical axis and the map distance is shown on the horizontal axis. The list of SNPs genotyped in the region is provided in the supplementary Table 4.

Linkage refinement analysis of the chromosome 4 BHR locus. Thirty-six single nucleotide polymorphism (SNP) markers were genotyped in the GAIN collection and the data was analyzed by multipoint nonparametric linkage methods. LOD scores and information content are shown in the vertical axis and the map distance is shown on the horizontal axis. The list of SNPs genotyped in the region is provided in the supplementary Table 5.
Discussion
Our genome scan revealed suggestive evidence of linkage to asthma and related phenotypes in nine chromosomal regions. There are several reports on linkage to the BHR phenotype and one of the asthma positional cloning reports is from a BHR locus.19 We also have a large collection of families with information on methacholine challenge test (184 sib-pairs with BHR in the genome scan set). Since asthma is a heterogeneous disease, examining specific mechanisms like BHR in an asthma family collection might increase the probability of success in identifying susceptibility genes. Three regions showing suggestive evidence of linkage to BHR were selected for follow-up studies. The linkage refinement analysis of the chromosome 2p locus was highly significant. The locus defined by one LOD drop interval spans 5 cM and the peak is centered on the SNP marker rs2063871. The flanking markers centromeric and telomeric to this region are rs737030 (80.16 cM) and rs920520 (69.2 cM), respectively. This region corresponds to a physical size of 12.2 Mb and the LOD score at this region reaches genome-wide significance level.39
Reports on genome-wide searches for linkage to asthma6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16 vary considerably, which could be due to differences in study design and sample size, as well as the fact that asthma is a heterogeneous disease with multifactorial etiology. Nevertheless, using recommended criteria,39 several regions had P-values that met the criteria for suggestive evidence of linkage (P≤0.002 or LOD≥1.9, reviewed in Hoffjan and Ober40). There are five reports where linkages to asthma or asthma-associated phenotypes reach genome-wide significance. They are on chromosome 2p for BHR (P=0.00002),41 chromosome 5q for mite sensitive asthma (P=1.3 × 10−7),42 chromosome 7q for total IgE (LOD 3.36),11 chromosome 11q for skin test index (P=0.00005)6 and chromosome 14 q for asthma (LOD 4).12 The susceptibility locus for IgE on 7q reported in the Finnish families has been positionally cloned recently.22 In addition, three recent reports describe the positional cloning of asthma genes on chromosome 20p13,19 chromosome 2q1421 and chromosome 13q14.20 We did not find evidence for linkage on chromosome 7q, 2q and 13q, respectively, where gene identification has already been reported. However, we found suggestive evidence for linkage on chromosome 20p near ADAM33 locus19 for strict asthma.
Two regions with significant evidence for linkage and seven regions with suggestive evidence were identified on our study. Although some of the regions with suggestive evidence for linkage may harbor asthma susceptibility genes, some of them may be the result of random fluctuation and Type I error. Comparison of these results with published genome scans will provide us with evidence of replication across the different studies. A summary of the overlap of our linkage to asthma and related phenotypes compared to the literature is shown in Table 4. There is evidence of replication of linkages on chromosome 1q, 2p, 3q, 4q, 12q, 20p and 21, but the linkages on chromosome 4p and 6q appear to be novel.
The identification of genes for complex diseases is hampered by genetic heterogeneity, epistatic interactions and gene–environment interactions. Though the population described in this report is from Caucasian subjects, genetic heterogeneity is a concern since the families are collected from Europe, the US and Australia. To address this issue, we conducted an investigation of pair-wise population genetic distances between the populations. The results (not shown) suggested that it was reasonable to separate the Greek and Norwegian sites from the overall group. Thus, after our initial screen that pooled all families, we separated the families into three separate groups: Greece, Norway and Western European descent and repeated the linkage analysis. As expected, the linkage results are consistent with the pooled analysis at the loci reported here and there was more contribution to the LOD score by the families from Western European descent, with the exception of chromosome 12q where the evidence of linkage is predominantly from the Greek families. So it is reasonable to conclude that the results reported in this study are not affected by genetic heterogeneity.
Characterizing heterogeneity in clinical phenotypes and establishing a standardized scheme to evaluate the phenotypic variability is a challenge in studying complex diseases like asthma. The diverse clinical presentation of asthma symptoms and severity make it difficult to define the correct phenotype for genetic analysis. We collected detailed phenotypic information from these families and used four major phenotypic definitions, which capture some of the diverse asthma phenotypes. A comparison of the linkages to specific phenotypes in the published literature will be more meaningful than the comparison of linkage to one general phenotype. By applying this principle, we identified five loci with evidence of replication at the phenotypic level (1q, 2p, 3q, 12q and 20p, see Table 3 for details).
The most significant evidence for linkage in this study is for the BHR phenotype on chromosome 2 (LOD score 4.58). It is interesting to note that in a genome scan of 97 German families, suggestive evidence of linkage to BHR was observed on chromosome 2p9, 43 at the marker D2S2298, which is located at ∼68 cM. The locus for BHR that we identified on chromosome 2p (61–106 cM) overlaps with the locus described in the German genome scan. The evidence of linkage for the strict asthma phenotype also reached genome-wide significance (LOD 3.98 at 51.5 cM) in our study. A number of genes (∼75) are mapped in the 12.2 Mb region. Several of these genes may have a role in the pathophysiology of asthma. One interesting candidate among these genes is suppressor of cytokine signaling 5 (SOCS5). The SOCS family of proteins is composed of eight members characterized by the presence of an Src homology 2 domains and a C-terminal conserved domain called the SOCS box.44 The SOCS proteins are implicated in the control of the balance between Th1 and Th2 cells. SOCS3 is predominantly expressed in Th2 cells and inhibits Th1 differentiation; conversely, SOCS5 is expressed predominantly in Th1 cells and inhibits Th2 differentiation.45 A high-density association study to identify the asthma susceptibility gene in this locus is underway.
Evidence of linkage for atopic asthma was previously reported at chromosome 1q and 3q. Linkage to mite sensitization (1q) at the marker D1S235 is just outside the boundary of our atopic asthma peak.46 Linkage to asthma was reported on chromosome 3q22 at marker D3S1744 with an MLS score of 2.41 for IgE and 1.29 for asthma.14 The same region (3q21) has also been shown to be linked to atopic dermatitis with an NPL score of 4.31.47 Along with asthma and allergic rhinitis, atopic dermatitis is an important manifestation of atopy and there is tremendous overlap in these phenotypes.48 In a recent report, significant evidence of linkage for mite sensitization was identified on chromosome 3q21.3 near the marker D3S3606 (MOD score 4.51).49 It is possible that the 3q region harbors genes related to allergic sensitization.
Using quantitative scores as phenotypic variables, linkage to asthma score was found on chromosome 12q near the marker D12S97.50 The same group also identified evidence of linkage to asthma affection status near marker D12S324. The suggestive evidence of linkage to asthma in our study is in the same region on chromosome 12.
The region on chromosome 20p we report here with a LOD score of 1.7 for strict asthma, maps near the ADAM33 locus reported for BHR.19 In that study, they found significant evidence of linkage to BHR (LOD 3.93) and subsequently identified the susceptibility gene. We used a strict definition of BHR (PC20≤8 mg), while our definition of strict asthma is similar to the definition of BHR in the ADAM 33 publication, which showed suggestive evidence of linkage. We defined strict asthma as having a positive methacholine test at the highest dose (16 mg) or a positive bronchodilator test and two or more clinical symptoms of asthma. Although there are overlaps in the linkage results, the role of ADAM33 to explain this linkage is not clear and further studies are necessary to asses the role of ADAM33 in our population.
In summary, we undertook a strict ascertainment protocol and the phenotype definitions were identical across the clinical centers. We also evaluated the reliability of asthma diagnosis across the different centers and demonstrated that the diagnosis is highly consistent. This collection spans three continents and represents one of the largest Caucasian asthma family collections. A single set of markers with common internal standards was used, providing high-quality marker data where the allele calls were consistent. Preliminary analysis of the genome scan data shows highly significant linkage of asthma to markers on chromosome 2p. Chromosome 2p contains many genes that could contribute to susceptibility to asthma. The size of the region indicates that a high-density association study using SNP markers is a good strategy to identify the asthma susceptibility genes from this locus. We conclude that we have mapped a high-risk asthma locus on chromosome 2p.
References
Jarvis D, Burney P : ABC of allergies: the epidemiology of allergic disease. BMJ 1998; 316: 607–610.
Jarvis D, Burney P : The epidemiology of allergic diseases. Br Med J 1988; 316: 607–609.
Smith DH, Malone DC, Lawson KA, Okamot LJ, Battista C, Saunders WB : A national estimate of the economic costs of asthma. Am J Resp Crit Care Med 1997; 156: 787–793.
Boushey HA, Holtzman MJ, Sheller JR, Nadel JA : Bronchial hyper-reactivity. Am Rev Resp Dis 1980; 12: 389–413.
Cookson WO, Moffatt MF : Genetics of asthma and allergic disease. Hum Mol Genet 2000; 9: 2359–2364.
Daniels SE, Bhattacharrya S, James A et al: A genome-wide search for quantitative trait loci underlying asthma. Nature 1996; 383: 247–250.
Xu J, Meyers DA, Ober C et al: Genomewide screen and identification of gene–gene interactions for asthma-susceptibility loci in three U.S. populations: collaborative study on the genetics of asthma. Am J Hum Genet 2001; 68: 1437–1446.
Ober C, Tsalenko A, Parry R, Cox NJ : A second-generation genomewide screen for asthma-susceptibility alleles in a founder population. Am J Hum Genet 2000; 67: 1154–1162.
Wjst M, Fischer G, Immervoll T et al: A genome-wide search for linkage to asthma. German Asthma Genetics Group. Genomics 1999; 58: 1–8.
Dizier MH, Besse-Schmittler C, Guilloud-Bataille M et al: Genome screen for asthma and related phenotypes in the French EGEA study. Am J Resp Crit Care Med 2000; 162: 1812–1818.
Laitinen T, Daly MJ, Rioux JD et al: A susceptibility locus for asthma-related traits on chromosome 7 revealed by genome-wide scan in a founder population. Nat Genet 2001; 28: 87–91.
Hakonarson H, Bjornsdottir US, Halapi E et al: A major susceptibility gene for asthma maps to chromosome 14q24. Am J Hum Genet 2002; 71: 483–491.
Koppelman GH, Stine OC, Xu J et al: Genome-wide search for atopy susceptibility genes in Dutch families with asthma. J Allergy Clin Immunol 2002; 109: 498–506.
Haagerup A, Bjerke T, Schiotz PO, Binderup HG, Dahl R, Kruse TA : Asthma and atopy – a total genome scan for susceptibility genes. Allergy 2002; 57: 680–686.
Hizawa N, Collins G, Rafnar T et al: Linkage analysis of Dermatophagoides pteronyssinus-specific IgE responsiveness with polymorphic markers on chromosome 6p21 (HLA-D region) in Caucasian families by the transmission/disequilibrium test. Collaborative Study on the Genetics of Asthma (CSGA). J Allergy Clin Immunol 1998; 102: 443–448.
Anonymous: A genome-wide search for asthma susceptibility loci in ethnically diverse populations. The Collaborative Study on the Genetics of Asthma (CSGA). Nat Genet 1997; 15: 389–392.
Cookson WO : A new gene for asthma: would you ADAM and Eve it? Trends Genet 2003; 19: 169–172.
Wills-Karp M, Ewart SL : Time to draw breath: asthma-susceptibility genes are identified. Nature 2004; 5: 376–387.
Van Eerdewegh P, Little RD, Dupuis J et al: Association of the ADAM33 gene with asthma and bronchial hyperresponsiveness. Nature 2002; 418: 426–430.
Zhang Y, Leaves NI, Anderson GG et al: Positional cloning of a quantitative trait locus on chromosome 13q14 that influences immunoglobulin E levels and asthma. Nat Genet 2003; 34: 181–186.
Allen M, Heinzmann A, Noguchi E et al: Positional cloning of a novel gene influencing asthma from chromosome 2q14. Nat Genet 2003; 35: 258–263.
Laitinen T, Polvi A, Rydman P et al: Characterization of a common susceptibility locus for asthma-related traits. Science 2004; 304: 300–304.
Suarez BK, Hampe CL, Van Eeerdeweigh P : Problems of replicating linkage claims in psychiatry; in: Gershon ES, Cloninger CR (eds):: Genetic Approaches to Mental Disorders. Washington, DC: American Psychiatric Association, 1994.
Helms PJ, Anderson W, Barnes K et al: Genetics Of Asthma International Network. Characterization of asthma, atopy and associated phenotypes in families identified through child probands with physician diagnosed asthma. Am J Hum Genet 2000; 67: 116.
Anonymous: Lung function testing: selection of reference values and interpretative strategies. American Thoracic Society [see comment]. Am Rev Resp Dis 1991; 144: 1202–1218.
Anonymous: Standards for the diagnosis and care of patients with chronic obstructive pulmonary disease (COPD) and asthma. This official statement of the American Thoracic Society was adopted by the ATS Board of Directors, November 1986. Am Rev Resp Dis 1987; 136: 225–244.
Juniper EF, Syty-Golda M, Hargreave FE : Histamine inhalation tests: inhalation of aerosol via a facemask versus a valve box with mouthpiece. Thorax 1984; 39: 556–557.
Cockcroft DW, Killian DN, Mellon JJ, Hargreave FE : Bronchial reactivity to inhaled histamine: a method and clinical survey. Clin Allergy 1977; 7: 235–243.
Barnes KC, Freidhoff LR, Horowitz EM et al: Physician-derived asthma diagnoses made on the basis of questionnaire data are in good agreement with interview-based diagnoses and are not affected by objective tests. J Allergy Clin Immunol 1999; 104: 791–796.
Ober C, Cox NJ, Abney M et al: Genome-wide search for asthma susceptibility loci in a founder population. The Collaborative Study on the Genetics of Asthma. Hum Mol Genet 1998; 7: 1393–1398.
Hauser ER, Crossman DC, Granger CB et al: A genomewide scan for early-onset coronary artery disease in 438 families: the GENECARD Study. Am J Hum Genet 2004; 75: 436–447.
Epstein MP, Duren WL, Boehnke M : Improved inference of relationship for pairs of individuals. Am J Hum Genet 2000; 67: 1219–1231.
McPeek MS, Sun L : Statistical tests for detection of misspecified relationships by use of genome-screen data. Am J Hum Genet 2000; 66: 1076–1094.
Cohen J : A coefficient of agreement for nominal scales. Educat Psychol Measur 1960; 20: 37–46.
Abecasis GR, Cherny SS, Cookson WO, Cardon LR : Merlin – rapid analysis of dense genetic maps using sparse gene flow trees [see comment]. Nat Genet 2002; 30: 97–101.
Kong A, Cox NJ : Allele-sharing models: LOD scores and accurate linkage tests. Am J Hum Genet 1997; 61: 1179–1788.
Huang Q, Shete S, Amos CI : Ignoring linkage disequilibrium among tightly linked markers induces false-positive evidence of linkage for affected sib pair analysis. Am J Hum Genet 2004; 75: 1106–1112.
Schaid DJ, McDonnell SK, Wang L, Cunningham JM, Thibodeau SN : Caution on pedigree haplotype inference with software that assumes linkage equilibrium. Am J Hum Genet 2002; 71: 992–995.
Lander E, Kruglyak L : Genetic dissection of complex traits: guidelines for interpreting and reporting linkage results [see comment]. Nat Genet 1995; 11: 241–247.
Hoffjan S, Ober C : Present status on the genetic studies of asthma. Curr Opin Immunol 2002; 14: 709–717.
Xu X, Fang Z, Wang B et al: A genomewide search for quantitative-trait loci underlying asthma [erratum appears in Am J Hum Genet 2002 Jul;71(1):215]. Am J Hum Genet 2001; 69: 1271–1277.
Yokouchi Y, Nukaga Y, Shibasaki M et al: Significant evidence for linkage of mite-sensitive childhood asthma to chromosome 5q31–q33 near the interleukin 12 B locus by a genome-wide search in Japanese families. Genomics 2000; 66: 152–160.
Immervoll T, Loesgen S, Dutsch G et al: Fine mapping and single nucleotide polymorphism association results of candidate genes for asthma and related phenotypes. Hum Mutat 2001; 18: 327–336.
Nicola NA, Nicholson SE, Metcalf D et al: Negative regulation of cytokine signaling by the SOCS proteins [Review] [43 refs]. Cold Spring Harbor Symp Quant Biol 1999; 64: 397–404.
Inoue H, Kubo M : SOCS proteins in T helper cell differentiation: implications for allergic disorders? Expert Rev Mol Med 2004; 6: 1–11.
Kurz T, Strauch K, Heinzmann A et al: A European study on the genetics of mite sensitization. J Allergy Clin Immunol 2000; 106: 925–932.
Lee YA, Wahn U, Kehrt R et al: A major susceptibility locus for atopic dermatitis maps to chromosome 3q21. Nat Genet 2000; 26: 470–473.
Cox HE, Moffatt MF, Faux JA et al: Association of atopic dermatitis to the beta subunit of the high affinity immunoglobulin E receptor. Br J Dermatol 1998; 138: 182–187.
Kurz T, Altmueller J, Strauch K et al: A genome-wide screen on the genetics of atopy in a multiethnic European population reveals a major atopy locus on chromosome 3q21.3. Allergy 2005; 60: 192–199.
Wilkinson J, Grimley S, Collins A, Thomas NS, Holgate ST, Morton N : Linkage of asthma to markers on chromosome 12 in a sample of 240 families using quantitative phenotype scores. Genomics 1998; 53: 251–259.
Haagerup A, Bjerke T, Schoitz PO, Binderup HG, Dahl R, Kruse TA : Allergic rhinitis – a total genome-scan for susceptibility genes suggests a locus on chromosome 4q24–q27. Eur J Hum Genet 2001; 9: 945–952.
Bouzigon E, Dizier MH, Krahenbuhl C et al: Clustering patterns of LOD scores for asthma-related phenotypes revealed by a genome-wide screen in 295 French EGEA families. Hum Mol Genet 2005; 15: 103–113.
Acknowledgements
We gratefully acknowledge the assistance of Mark Hall, Steven Henry, Rodney Winkler and Michael Stubbins of GlaxoSmithKline for data management support, and Shahid Bhatti and Tejinder Bhinder of GlaxoSmithKline for their assistance with SNP identification and genotyping.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on European Journal of Human Genetics website (http://www.nature.com/ejhg).
Supplementary information
Rights and permissions
About this article
Cite this article
Pillai, S., Chiano, M., White, N. et al. A genome-wide search for linkage to asthma phenotypes in the genetics of asthma international network families: evidence for a major susceptibility locus on chromosome 2p. Eur J Hum Genet 14, 307–316 (2006). https://doi.org/10.1038/sj.ejhg.5201532
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.ejhg.5201532
Keywords
This article is cited by
-
Meta-analysis of 20 genome-wide linkage studies evidenced new regions linked to asthma and atopy
European Journal of Human Genetics (2010)
-
Significant evidence for linkage to chromosome 5q13 in a genome-wide scan for asthma in an extended pedigree resource
European Journal of Human Genetics (2009)
-
Meta-analysis of genome-wide linkage studies of asthma and related traits
Respiratory Research (2008)
-
Interleukin 18 receptor 1 gene polymorphisms are associated with asthma
European Journal of Human Genetics (2008)
-
Polymorphisms in the endothelin-1 (EDN1) are associated with asthma in two populations
Genes & Immunity (2008)
