
Abstract
We have identified a girl with characteristic features of Rett syndrome (RTT) who carries a de novo balanced translocation involving chromosomes 1 and 7. Both breakpoints were mapped by fluorescence in situ hybridization with selected genomic clones from the regions of interest. Southern blot hybridisations, utilizing probes derived from breakpoint spanning BACs, detected several aberrant fragments specific for the patient. Sequence analysis of the cloned junction fragment indicated that on chromosome 1 the predominantly brain-expressed Netrin G1 (NTNG1) gene is disrupted, whereas on chromosome 7 there was no indication for a truncated gene. The chromosome 1 breakpoint lies within the 3′ part of NTNG1 and affects alternatively spliced transcripts, suggesting that the phenotype in this patient is the result of disturbed NTNG1 expression. In silico translation of the NTNG1 splice variants predicted protein isoforms with different C-termini: one membrane bound through a glycosylphosphatidylinositol anchor and the other soluble. The membrane-bound protein isoform would be affected by the breakpoint, whereas the soluble form would remain intact. Our results suggest that the central nervous system is sensitive to NTNG1 expression levels and that NTNG1 is a novel candidate disease gene for RTT.
Similar content being viewed by others


Introduction
Breakpoint mapping in patients with apparently balanced disease-associated chromosome rearrangements (DBCRs) has been instrumental in identifying disease genes. In order to find new genes involved in mental retardation, we have embarked on a systematic study of X chromosomal and autosomal DBCRs in patients with cognitive deficits. In this context, we have investigated a girl with Rett syndrome (RTT) and a de novo balanced chromosome rearrangement. RTT, a progressive neurodevelopmental disorder, is a leading cause of neurological dysfunction in female subjects. RTT has been considered as a monogenic disorder that is caused by defects in MECP2.1, 2 Indeed, mutations in MECP2 have been identified in up to 90% of patients with classic RTT. However, both the lack of MECP2 mutations in a small group of classic RTT cases and the low frequency of MECP2 mutations in atypical RTT patients suggest that other gene defects may also play a role in this disorder. The existence of locus heterogeneity is supported by the recent identification of mutations in the CDKL5 gene.3, 4, 5 Here, we describe a patient with RTT and a balanced translocation, which disrupts an autosomal gene that is expressed in the brain.
Materials and methods
Cytogenetic studies
A stained chromosome suspension was used to collect the normal and the derivative chromosomes 1 and 7, by bivariate flow sorting using a FACStar Plus cell sorter. Whole chromosome paints were prepared by degenerate oligonucleotide primed PCR on flow-sorted chromosomes.
A cross-species colour paint using gibbon chromosomes and a 24-colour paint probe using five human chromosome pools were both prepared in-house. The telomeric regions of the chromosomes were screened with the Chromoprobe Multiprobe–T System from Cytocell. YAC, BAC and PAC clones used for breakpoint mapping were made into FISH probes as described by Wirth et al.6
FISH with the Angelman/Prader–Willi chromosome region probe E24 did not reveal a deletion at 15q11.2. DNA methylation studies for microdeletion/uniparental disomy on chromosome 15 for Angelman syndrome were also negative.
MECP2 and CDKL5 mutation analysis
In view of the diagnosis, we performed FISH using a PAC clone containing MECP2. Screening for mutations in MECP2 was done by sequencing PCR-amplified exons and for CDKL5 by DHPLC analysis. PCR primers and DHPLC conditions are available on request. Furthermore, quantitative PCR, extending to the promoter region, exon 1 and exon 2 of MECP2, was carried out as recently described.7
cDNA synthesis and RT-PCR
Total patient and control RNA isolated from lymphoblastoid cell lines and fibroblast cell lines was used for reverse transcription. Human HPRT primers assessed the quality of the cDNA. Semiquantitative RT-PCRs, which amplify both transcript variants, were carried out by using primers located in exon 4 (5′-AAC ATG GCA GCC CTA TCA GT-3′) and in exon 5 (5′-GTA GTG TTG TGC TCA CAT TC-3′) and a QuantumRNA Classic 18S Internal Standards kit (Ambion). Semiquantitative RT-PCRs that detect either variant 1 or 2 were performed with the QuantumRNA Classic II 18S Internal Standards kit (Ambion). For variant 2, specific primers located in exon 7 (5′-GAA TGT CTG CGA CAA CGA GC-3′, 5′-CAC AAT ATG CTC TGC AGC TG-3′) were used. Furthermore, amplifications of only the transcript variant 1 were with primers located in exon 2 (5′-ACC TGT CAA AGT CAC TGA TC-3′) and in exon 3 (5′-GAG TGA TCC TCA ATG TTC AC-3′).
RT-PCR for expression analysis of EST BX643283 on chromosome 7 was performed on cDNA generated from fetal brain, lymphoblastoid and fibroblast RNA. PCR reaction was carried out with primers 5′-CCA TGC CTT GCT TGT CCA CA-3′ and 5′-GAA CAA GGC GGA GCT GAA CA-3′. As a control for cDNA quality, we have used the above-mentioned primers specific for NTNG1 variant 2.
Netrin G1 (NTNG1) mutation search
For NTNG1 mutation search, we amplified all coding exons from genomic patient DNA by PCR with primers located in flanking introns. Primer sequences are available on request. PCR products were analysed by DHPLC with the WAVE system (Transgenomic).
In silico analysis of NTNG1
We performed in silico analysis for the predicted NTNG1 proteins concerning the possibility of a potential cleavage site for glycosylphosphatidylinositol (GPI) anchoring following the ω+2 rule,8 by using the DGPI website available at ExPASy Proteomics tools (www.expasy.org/tools/).
Results
Clinical history
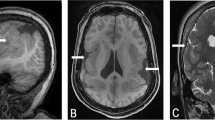
Pregnancy was uneventful and the patient was born at term by normal vaginal delivery. Birth weight was normal and head circumference (OFC) was at the 90th centile. Development was normal until the age of 5 months at which time she started to have ‘eye rolling’ episodes. By 6 months, she also began finger-feeding herself. At 7 months, she was hospitalized following a prolonged afebrile convulsion. A CAT scan of the brain during this hospital admission suggested a degree of atrophy and this finding was also confirmed on an MRI scan. Chromosome analysis revealed a de novo apparently balanced translocation 46,XX,t(1;7)(p22;q32). At a follow-up examination at 8.5 months of age, the child's OFC was 45.5 cm (just below the 90th centile) and weight was at 7.9 kg (just below the 50th centile). She was still experiencing frequent ‘eye rolling’ episodes, now being considered as seizures, and anticonvulsive treatment was commenced. She was able to fix and follow with her eyes but attention was poorly sustained and she had rather limited social response. She did not sit unsupported, had some head lag and was unable to take weight on her legs though power and tone in the lower limbs were apparently normal. A biotinidase assay and visual evoked potentials were normal. A sleep deprived EEG performed at the age of 2 years showed a rhythmic background with high amplitude bursts facilitated by sleep. Evaluation of a series of CT and MRI scans of the brain taken until the age of 2.5 years showed a slight delay in the rate of myelination as the only abnormality. The widened subarachnoid space anteriorly, which had suggested cerebral atrophy in earlier films, had largely resolved. By the age of 3.5 years, the epileptic seizures had been brought under control with Clobazam and Lamotrigine. At 4 years of age, there was concern that the enlarged tonsils were the cause of her recurrent chest infections, which necessitated hospitalization, and of her total refusal of food and drink on occasions. Her mother had to administer fluids via a nasogastric tube when the child went through a period of total refusal. A hearing test was also conducted at the time and this was reported as normal.
When we saw her 2 months later, the mother reported that the child's main problems were her abnormal sleep pattern and refusal of oral fluids. She tended to grind her teeth and to hum all the time. She was not toilet trained and was doubly incontinent. She walked with a wide-based unsteady gait with arms flexed at the elbow. She had marked hand stereotypy of the clasping, mouthing and wringing type, which had been present since the age of 2 years (Figure 1). She was able to maintain some eye contact but would not socialize. She had no speech but had said ‘mum’ and ‘dad’ at 13 months of age. Her growth was below the 3rd centile for height and weight and the OFC was along the 25th centile. Minor dysmorphic features included full cheeks, full lips, a bulbous nasal tip and clinodactyly of the 5th finger, but she was not dysmorphic overall. The rest of the physical examination was normal. As the current criteria for the diagnosis of RTT were fulfilled,9 a firm diagnosis of the condition was made.

Clinical photos of the patient at the age of 4 years 2 months. Note stereotypic hand movements. A nasogastric tube is in situ and is plastered to her right cheek.
Molecular cytogenetic analyses
Subtelomeric FISH confirmed the translocation between chromosomes 1 and 7 and the presence of all telomeres (Figure 2a, b). Following forward and reverse painting (Figure 2d, f and data not shown), it was suspected that the breakpoint on 1p was more proximal than was reported by GTG-banding; this was confirmed by FISH with YAC probes. The karyotype was redefined as 46,XX,t(1;7)(p13.3;q31.3) by BAC FISH.

Molecular and conventional cytogenetic studies. (a, b) FISH using Chromoprobe Multiprobe–T System from Cytocell. Partial metaphase spreads showing the balanced translocation between 1p (green signals in a) and 7q (red signals in b). The interphase nucleus in (b) shows two red and two green signals, representing the telomeres of chromosomes 1 and 7. (c, e) G-banded normal and derivative chromosomes 1 and 7. Translocation breakpoints are indicated. (d, f) FISH using normal chromosome 1 (green) and 7 (red) whole chromosome painting probes, on patient's chromosomes. (g) FISH with the breakpoint spanning BAC clone RP11-436H6 (Accession number AL513187). Signals are seen on normal chromosome 1, der(1) and der(7).
On chromosome 7, PAC clone RP5-865F17 (Accession number AC006010) gave signals proximal to the breakpoint and clone RP5-902E20 (Accession number AC006148) showed signals distal to the breakpoint (data not shown).
FISH mapping of the chromosome 1 breakpoint with BAC clone RP11-436H6 (Accession number AL513187) showed split signals on both derivative chromosomes (Figure 2g). The partially sequence overlapping clones RP11-24J14 (Accession number AC016085) and RP11-270C12 (Accession number AC114491) gave split signals in four out of 14 metaphases and signals proximal to the breakpoint in the other metaphases (data not shown).
The rearrangement disrupts the NTNG1 gene on chromosome 1
The breakpoint spanning clones on chromosome 1 contain exons of the NTNG1 gene, suggesting that this gene is disrupted by the breakpoint. Southern blot hybridizations of digested DNAs, with a probe encompassing the region between 186 651 and 187 217 bp on the breakpoint spanning BAC RP11-270C12, showed aberrant fragments in the patient DNA but not in the control (Figure 3a). This result located the breakpoint within a region of 568 bp on chromosome 1. To determine the position of the breakpoint on chromosome 7, we cloned the junction fragment from derivative chromosome 7. Using DraI-digested patient DNA for PCR amplifications, we obtained two PCR fragments. The smaller PCR fragment was specific for the patient (Figure 3b) and is composed of sequence derived from chromosome 1 and chromosome 7 with three additional nucleotides inserted at the breakpoint site (Figure 3c). The breakpoint is located in the genomic region of 107 686 151 bp on chromosome 1 and 123 365 462 bp on chromosome 7 (UCSC hg17 assembly, May 2004) [see also Supplementary Information].

Molecular characterization of the translocation breakpoints. (a) Southern blot of patient (P) and control (C) digested genomic DNA hybridized with a PCR product corresponding to the region 186 650–187 217 bp of the chromosome 1 breakpoint spanning BAC clone RP11-270C12. Aberrant fragments are seen in patient DNA and are not present in control DNA. (b) Suppression PCR on DraI digested patient (P) and control (C) DNA. Lanes 2 and 4 are negative controls. (c) PCR and sequencing of the breakpoints showing nucleotides from der(7) and der(1) together with an insertion of three nucleotides at the der(7) breakpoint and an extra two nucleotides at the der(1) breakpoint.
At least two different NTNG1 splice variants exist, represented by GenBank Accession numbers AB023193 and BC030220, and are designated variant 1 and variant 2, respectively. The RNAs differ in their first exons and in their 3′ ends. Different 3′ ends are ascribed to an alternatively used splice site in exon 6 (Figure 4). The breakpoint on chromosome 1 lies within intron 6, indicating that NTNG1 transcript variant 2 is directly affected by the translocation, whereas the shorter variant 1 is likely unaffected by the breakpoint.

Expression analysis of NTNG1 in the patient. On the left are schematic diagrams (not to scale) depicting the exon–intron structure of NTNG1 and the two major transcript variants; on the right are the results for the semiquantitative RT-PCRs; upper row shows amplification of both NTNG1 transcript variants in patient (P) and controls (C); second and third rows show the results specific for each transcript variant. All PCR products were compared to 18S internal standards.
In contrast, no known gene is disrupted by the chromosome 7 breakpoint. In silico sequence analysis revealed that the sequence of chromosome 7 obtained by breakpoint cloning is located on PAC clone RP5-865F17 (Accession number AC006010). There is only a single spliced EST (Accession number BX643283) in this region. Repeated RT-PCR experiments with fetal brain, lymphoblastoid and fibroblast cDNAs failed to produce a product.
Expression analysis of NTNG1 in the patient with t(1;7) translocation
To examine the impact of the chromosome rearrangement on NTNG1 expression, we have studied the expression level of the different splice variants in the patient cell line. By semiquantitative RT-PCR experiments, we could show that variant 2 (Accession number BC030220), which is truncated due to the chromosomal rearrangement, is less abundant in the patient RNA compared to control RNAs (Figure 4, right-hand side, lowest panel). In contrast, transcript variant 1 (Accession number AB023193), which is not truncated by the breakpoint, is present at a comparable level in the patient and controls (Figure 4, right-hand side, middle panel). These findings are supported by the results of another set of RT-PCR experiments using primers located in exons 4 and 5, which amplify all NTNG1 splice variants. As can be seen in Figure 4 (right-hand side, upper panel), NTNG1 expression is significantly lower in the patient compared to controls. Comparison of all three sets of RT-PCR experiments suggests that most likely, the disrupted variant 2 is unstable and degraded. The remaining expression of this variant probably comes from the normal copy of chromosome 1. In addition, the unaltered expression of variant 1 indicates that no downstream transcriptional regulator for this variant exists, which had otherwise been separated from this transcription unit through the rearrangement event.
Mutation analysis of NTNG1 in a panel of patients with typical and atypical RTT
After having found that one of the NTNG1 transcript variants is truncated by the chromosome 1 breakpoint, we screened the entire NTNG1 coding sequence and neighbouring intronic sequences for mutations in a small group of 30 typical and atypical RTT patients with no mutation in MECP2 or CDKL5. This mutation search was negative.
Discussion
Subsequent to breakpoint mapping in a patient with classic RTT associated with a de novo translocation and an intact MECP2 and CDKL5, we have searched for possible disease genes in the affected chromosome regions. On chromosome 7, the rearrangement disrupted a putative EST. Repeated attempts failed to verify this transcript using RNA from different cell types, suggesting that this EST is either not expressed in fetal brain, or its quality is questionable. Therefore, it is rather unlikely that the chromosome 7 breakpoint plays a role in the disease phenotype, although we cannot exclude an effect on a gene located in the vicinity of the breakpoint.
The more appropriate candidate disease gene is NTNG1, which is directly truncated by the chromosome 1 breakpoint. We propose that the patient's phenotype is most likely the result of disturbed NTNG1 function.
NTNG1 transcripts are alternatively spliced and only the longer RNA variants are truncated by the breakpoint, suggesting that haploinsufficiency of a single NTNG1 splice variant is associated with RTT in our patient, and that proper NTNG1 function is critical for normal development.
Absence of NTNG1 mutations in a small number of other RTT patients may indicate that mutations in this gene may be rare, or that only a small spectrum of all possible mutations produce an RTT phenotype. Mutation screening in a larger group of patients will shed more light on the impact of NTNG1 on RTT.
Netrin G1, also known as laminet-1, was initially identified as a new member of the small UNC6/netrin family in mice. However, unlike classical netrins, human and mouse NTNG1 are alternatively spliced. Likewise, and currently only established for the mouse, Ntng1 is not expressed in the ventral midline of the CNS and consists of at least six different protein isoforms of which five isoforms are predominantly linked to the plasma membrane via carboxy-terminal GPI lipid anchors. Furthermore, Ntng1 proteins lack any appreciable affinity to receptors for classical netrins.10 Ntng1 transcripts are most abundant in the CNS with highest expression in the dorsal thalamus, olfactory bulb, and inferior colliculus. Interestingly, Ntng1 is also expressed in the cerebellar inferior olive nuclei, and thus may play a role in forming functional circuits for controlling motor activities. Immunohistochemical studies indicated that Ntng1 protein is present on thalamocortical but not corticothalamic axons and it has been proposed to provide short-range cues for axonal and/or dendritic behaviour.10, 11, 12 However, these studies do not distinguish between different splice variants.
Our in silico analysis for human NTNG1 proteins revealed that at least one protein isoform (Accession number Q8N633) harbours a ω-site for GPI anchoring in its C-terminal part. In the patient, the sequence that codes for this cleavage site is separated from NTNG1 through the chromosome rearrangement. Although our results obtained by RT-PCR suggest lowered NTNG1 expression, there is also the possibility that truncated NTNG1 transcripts are translated into protein and that these proteins interfere with the activity of the wild-type form resulting in a dominant negative effect. In this context, it is noteworthy that a recently identified ligand for Ntng1, NGL-1, stimulates neurite outgrowth from the thalamic neurons when bound to a surface receptor present on thalamocortical axons. Removal of GPI-anchored proteins from the cell surface severely abrogates neurite outgrowth, but can be partially restored in the presence of a high concentration of soluble Ntng1.13
The shorter NTNG1 splice variant is not affected by the breakpoint and therefore is expressed at a normal level. The corresponding protein has no predicted ω-site and it is expected that no GPI moiety is attached. This makes it highly likely that, as in mice, in humans both types of NTNG1 protein exist, that is a membrane bound form and a soluble form, which possibly act, respectively, as short- and long-range acting axon guidance cues. For the repulsive GPI-anchored axon guidance molecule RGMa, involved in proper innervations of the hippocampus and outgrowth of entorhinal axons, it has been shown that separation from its GPI anchor results in abnormal innervations, whereas the long-range outgrowth is not affected.14 If a similar situation exists for Netrin G1, we could expect that local axonal wiring and short-range axon guidance are disturbed in the patient, whereas the long-range acting axon guidance cues would be intact.
From different investigations it has been concluded that RTT may be a disorder due to an alteration of neuronal plasticity and due to an arrest of neuronal development. This is also suggested by the findings on brain scans of our patient that were taken at 7 months of age. In the RTT brain, fewer neurons express MeCP2 than in the normal brain. This reduction is most apparent in two brain areas, the brain stem and the thalamus. The expression pattern of NTNG1 in the human brain is not known, but it is noteworthy that in the mouse, the thalamus is the structure with the most prominent Ntng1 expression. Given that disturbed NTNG1 expression in the translocation patient produces a phenotype of classic RTT, otherwise caused by a mutation in MECP2, it is possible that both molecules act in the same pathogenic pathway.
In genome-wide screens, proximal chromosome 1p has been implicated as the most likely location for an autism-predisposing locus.15, 16 Against this background, our findings may justify further studies to clarify the possible role of NTNG1 in the pathogenesis of autism.
Accession codes
Accessions
GenBank/EMBL/DDBJ
References
Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY : Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet 1999; 23: 185–188.
Renieri A, Meloni I, Longo I et al: Rett syndrome: the complex nature of a monogenic disease. J Mol Med 2003; 81: 346–354.
Tao J, Van Esch H, Hagedorn-Greiwe M et al: Mutations in the X-linked cyclin-dependent kinase-like 5 (CDKL5/STK9) gene are associated with severe neurodevelopmental retardation. Am J Hum Genet 2004; 75: 1149–1154.
Weaving LS, Christodoulou J, Williamson SL et al: Mutations of CDKL5 cause a severe neurodevelopmental disorder with infantile spasms and mental retardation. Am J Hum Genet 2004; 75: 1079–1093.
Scala E, Ariani F, Mari F et al: CDKL5/STK9 is mutated in Rett syndrome variant with infantile spasms. J Med Genet 2005; 42: 103–107.
Wirth J, Nothwang HG, van der Maarel S et al: Systematic characterisation of disease associated balanced chromosome rearrangements by FISH: cytogenetically and genetically anchored YACs identify microdeletions and candidate regions for mental retardation genes. J Med Genet 1999; 36: 271–278.
Laccone F, Junemann I, Whatley S et al: Large deletions of the MECP2 gene detected by gene dosage analysis in patients with Rett syndrome. Hum Mutat 2004; 23: 234–244.
Eisenhaber B, Bork P, Eisenhaber F : Sequence properties of GPI-anchored proteins near the omega-site: constraints for the polypeptide binding site of the putative transamidase. Protein Eng 1998; 11: 1155–1161.
Hagberg B, Hanefeld F, Percy A, Skjeldal O : An update on clinically applicable diagnostic criteria in Rett syndrome. Comments to Rett Syndrome Clinical Criteria Consensus Panel Satellite to European Paediatric Neurology Society Meeting, Baden Baden, Germany, 11 September 2001. Eur J Paediatr Neurol 2002; 6: 293–297.
Nakashiba T, Ikeda T, Nishimura S et al: Netrin-G1: a novel glycosyl phosphatidylinositol-linked mammalian netrin that is functionally divergent from classical netrins. J Neurosci 2000; 20: 6540–6550.
Nakashiba T, Nishimura S, Ikeda T, Itohara S : Complementary expression and neurite outgrowth activity of netrin-G subfamily members. Mech Dev 2002; 111: 47–60.
Yin Y, Miner JH, Sanes JR : Laminets: laminin- and netrin-related genes expressed in distinct neuronal subsets. Mol Cell Neurosci 2002; 19: 344–358.
Lin JC, Ho WH, Gurney A, Rosenthal A : The netrin-G1 ligand NGL-1 promotes the outgrowth of thalamocortical axons. Nat Neurosci 2003; 6: 1270–1276.
Brinks H, Conrad S, Vogt J et al: The repulsive guidance molecule RGMa is involved in the formation of afferent connections in the dentate gyrus. J Neurosci 2004; 24: 3862–3869.
Risch N, Spiker D, Lotspeich L et al: A genomic screen of autism: evidence for a multilocus etiology. Am J Hum Genet 1999; 65: 493–507.
Auranen M, Nieminen T, Majuri S, Vanhala R, Peltonen L, Jarvela I : Analysis of autism susceptibility gene loci on chromosomes 1p, 4p, 6q, 7q, 13q, 15q, 16p, 17q, 19q and 22q in Finnish multiplex families. Mol Psychiatry 2000; 5: 320–322.
Acknowledgements
We are grateful to the patients and their families. We thank Patricia CM O'Brien (CVBS, Cambridge) for performing chromosome sorting. This work was supported by the German Human Genome Program (01KW99087), the National Genome Research Network (01GR0105) and a scholarship to IB from the Association of Commonwealth Universities. Wilhelm Johannsen Centre for Functional Genome Research is established by the Danish National Research Foundation.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on European Journal of Human Genetics website (http://www.nature.com/ejhg)
Supplementary information
Rights and permissions
About this article
Cite this article
Borg, I., Freude, K., Kübart, S. et al. Disruption of Netrin G1 by a balanced chromosome translocation in a girl with Rett syndrome. Eur J Hum Genet 13, 921–927 (2005). https://doi.org/10.1038/sj.ejhg.5201429
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.ejhg.5201429
Keywords
This article is cited by
-
Homozygous frameshift variant in NTNG2, encoding a synaptic cell adhesion molecule, in individuals with developmental delay, hypotonia, and autistic features
neurogenetics (2019)
-
Convergence of independent DISC1 mutations on impaired neurite growth via decreased UNC5D expression
Translational Psychiatry (2018)
-
Rett Syndrome: Reaching for Clinical Trials
Neurotherapeutics (2015)
-
Custom oligonucleotide array-based CGH: a reliable diagnostic tool for detection of exonic copy-number changes in multiple targeted genes
European Journal of Human Genetics (2013)
-
De novo mutations in neurological and psychiatric disorders: effects, diagnosis and prevention
Genome Medicine (2012)
