
Abstract
Oculo-facio-cardio-dental (OFCD) syndrome is a rare X-linked dominant condition with male lethality characterized by microphthalmia, congenital cataracts, facial dysmorphic features, congenital heart defects, and dental anomalies. Mutations in BCOR (BCL6 co-repressor) located in Xp11.4 have been described to cause OFCD syndrome. Lenz microphthalmia syndrome is inherited in an X-linked recessive pattern comprising microphthalmia/anophthalmia, mental retardation, malformed ears, digital, skeletal, and urogenital anomalies (synonym: microphthalmia with associated anomalies (MAA)). One locus for MAA has been mapped to Xq27–q28. Nonetheless, linkage and subsequent mutation analysis revealed a single missense mutation (p.P85L) in BCOR in a large family with presumed Lenz microphthalmia syndrome (MAA2). We describe novel mutations in BCOR in three patients with OFCD syndrome, two small deletions (c.2488_2489delAG and c.3286delG) and a submicroscopic deletion of about 60 kb encompassing at least BCOR exons 2–15. No BCOR mutation was detected in eight patients with Lenz microphthalmia syndrome. Our data confirm that BCOR is the causative gene for OFCD syndrome; however, the failure to identify any mutation in patients with Lenz microphthalmia syndrome together with the oligosymptomatic phenotype in the reported MAA2 patients suggest that BCOR is not the major gene for this syndrome.
Similar content being viewed by others


Introduction
Oculo-facio-cardio-dental syndrome (OFCD syndrome; OMIM 300166) is inherited as an X-linked dominant condition with presumed male lethality and comprises congenital cataracts, microphthalmia, dysmorphic craniofacial features, congenital heart defects, and dental anomalies.1, 2 Recently, mutations in BCOR, encoding the BCL6 co-repressor, have been described in five sporadic and two familial cases with OFCD syndrome.3 Ng et al3 proposed that due to the phenotypic overlap OFCD and Lenz microphthalmia syndrome are allelic disorders. Lenz microphthalmia syndrome (microphthalmia with associated anomalies (MAA); OMIM 309800) is an X-linked recessive condition comprising microphthalmia/anophthalmia with mental retardation, malformed ears, skeletal, renal, and urogenital anomalies. Two genetic loci that can cause what is currently considered to be Lenz microphthalmia syndrome have been reported, one located in Xq27–q28 (MAA)4 and the second in Xp11.4–p21.2 (MAA2).5 A single missense change, c.254C>T (p.P85L), in BCOR has been found in patients of a family with MAA2.3
We performed mutation analysis in one reported patient2 and two undescribed patients with OFCD syndrome, as well as in eight patients affected with Lenz microphthalmia syndrome, to confirm that mutations in BCOR are causative for both conditions.
Materials and methods
Subjects
Patients with OFCD syndrome
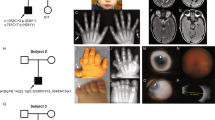
The clinical findings of three female patients with OFCD syndrome are summarized in Table 1. For these patients, the diagnosis of OFCD syndrome was established on the basis of a distinct pattern of eye, heart, dental, and craniofacial anomalies. In each case, family history was unremarkable. Clinical features of two affected individuals are shown in Figure 1a–c.

Photographs of patients 2 and 3 with OCFD syndrome. (a) Facial appearance of patient 2 at age of 3 years, note unilateral microphthalmia, laterally curved eyebrows, simple and long philtrum, and broad diastema. (b) Facial view of patient 3 at the age of 2.5 years showing bilateral microphthalmia, upward slanting palpebral fissures, mildly broad nasal tip, and long philtrum. (c) Broad hallux and camptodactyly of toes 2, 3 of patient 2.
Patients with Lenz microphthalmia syndrome
We have studied eight male patients with microphthalmia/anophthalmia, mental retardation, and a wide range of associated extraocular anomalies. In each case, the family history was unremarkable and chromosomal studies revealed a normal karyotype. Patients were diagnosed as having Lenz microphthalmia syndrome if at least the following features were present: microphthalmia/anophthalmia, microcephaly, mental retardation, and abnormalities of the extremities. To analyze a possible association of a broader clinical spectrum of syndromic microphthalmia/anophthalmia in males and the presence of BCOR mutations, we also studied patients presenting with at least the ocular phenotype, mental retardation, and additional abnormalities. The clinical findings of the patients are summarized in Table 2.
Microphthalmia/anophthalmia (8/8), mental retardation (8/8), microcephaly (7/7), ear anomalies (7/7), and short stature (6/8) were the most constant features in the patients. Abnormal teeth (4/5), and urogenital anomalies (4/8) seem to be frequently associated features. Craniofacial findings including short philtrum and high palate were only documented in a minority of them. Abnormalities of the extremities (syndactyly of hands and feet, hypoplastic terminal phalanges of thumbs) are features described in the original report6 and were found in 3/7 patients. The final diagnostic criteria for Lenz microphthalmia syndrome can only be delineated once the causative gene has been identified and mutation analysis can be performed.
Informed consent was obtained from all participants or their parents and the study was approved by the review board of the institutions of the primary care physicians.
Mutation and single-nucleotide polymorphisms (SNP) analysis
We amplified the coding region of BCOR (15 exons; GenBank accession no. AY316592), including the flanking intronic sequences, from genomic DNA that was isolated by standard procedures. Primer sequences and PCR conditions are available on request. PCR products were directly sequenced with the BigDye Terminator ready reaction kit (PE Applied Biosystems) on an ABI 377 Prism automated sequencer (PE Applied Biosystems).
For SNP analysis of the parents of patient 3 and the patient, we amplified various DNA fragments containing 19 SNPs in the BCOR gene (refSNP Ids: rs5963725, rs4393046, rs5963154, rs1388717, rs3810694, rs5917931, rs6520618, rs5917933, rs6610384, rs4076107, rs6520620, rs11797578, rs5963156, rs3810693, rs3749422, rs3840969, rs12007362, rs6609051, and rs4546795) and directly sequenced them.
Fluorescence in situ hybridization (FISH)
Metaphase spreads from peripheral blood lymphocytes or lymphoblastoid cells of patient 3 were made by standard procedure. Fosmid clones from library G248P8 (2501D4, 1422H2, 7601G5, 5351A7, 9225G6, 9478F10, 2316B5, and 5335D1) were obtained from BACPAC Resources (Oakland, USA). Fosmid DNA was isolated using Plasmid Midi kit (Qiagen) and labeled with biotin-16-dUTP (Roche) by nick translation (Roche). Fluorescein isothiocyanate (FITC) labeled streptavidin (1:200) was used for detection. The X centromere probe labeled by FITC (Appligene Oncor) was used to identify the X chromosome. Chromosomes were counterstained with DAPI. Slides were analyzed with a Leica fluorescence microscope equipped with Smart Capture Software (Vysis).
Results
Small deletions in BCOR in two patients with OFCD syndrome
We investigated three female patients, all presenting with OFCD syndrome. By PCR, we amplified all exons and flanking intronic sequences of the BCOR gene in the three patients. We identified two novel mutations in BCOR, a deletion of two nucleotides, c.2488_2489delAG (p.Ser830CysfsX5), in exon 4 of patient 2 and a single-nucleotide deletion, c.3286delG (p.Glu1096ArgfsX16), in exon 7 of patient 1 (Table 1). The mother of patient 2 does not carry the c.2488_2489delAG mutation. As neither the father of patient 2 nor the parents of patient 1 were available, we screened 100 X chromosomes for the presence of the two changes but did not detect them (data not shown). No obvious mutation, using these techniques, was discovered in patient 3.
Presence of a large deletion in one patient with OFCD syndrome
To search for a submicroscopic deletion in patient 3, we amplified 19 SNPs in the BCOR gene in the patient and her parents. Sequence analysis of the PCR products revealed that mother and father show different alleles for the two SNPs rs6520618 (exon 4) and rs5917931 (intron 6) (Figure 2a). Sequence analysis of the corresponding amplicons of patient 3 showed that she carries only the maternal allele for both variations (A+A) (Figure 2a), suggesting loss of the paternal allele due to partial deletion of the BCOR gene, including part of exon 4, intron 4 to exon 6, and part of intron 6.

Patient 3 carries a large deletion encompassing almost the entire BCOR gene. (a) SNP analysis suggested loss of the paternal BCOR allele in patient 3. A pedigree of patient 3 and her parents is shown on the top. Parts of the electropherograms of SNP rs6520618 (BCOR exon 4) and rs5917931 (BCOR intron 6) for patient 3 and her parents are presented below the pedigree. Variable nucleotides are underlined and indicated by arrows. Patient 3 carries only the maternal allele for both nucleotide variations. (b) Schematic representation of the genomic organization of the BCOR gene (GenBank Accession no. AY316592) and the locations of fosmid clones used in this study (not drawn to scale). At the top, BCOR exons are represented by black boxes and are partially numbered. The start codon is located in exon 2, whereas the stop codon is in exon 15. The 3′ untranslated region is indicated by a white box. The localization of the two informative SNPs in exon 4 and intron 6 is indicated by green arrowheads. Fosmid inserts are shown by lines and their respective names are given. Fosmid clones showing two signals in FISH are indicated by a black line, those producing only a single signal by a red line, and fosmids with a strong and a weak signal by a blue line. The size of the deletion is indicated by a black line below the fosmids; hatched lines indicate that the proximal and distal deletion boundaries have not been determined precisely. (c, d) FISH analysis with two fosmid clones hybridized to metaphase spreads of patient 3. (c) Fosmid 1422H2 is labeled by FITC (green) and produced a strong signal on one X chromosome and a weak signal on the second one, suggesting that the centromeric deletion breakpoint maps on the fosmid insert. (d) For fosmid 9225G6 (green), only a single signal on one X chromosome was detected. Chromosomes are counterstained with DAPI.
To investigate the presence of a submicroscopic deletion on one of the X chromosomes of patient 3, we performed FISH with various fosmid clones spanning the BCOR gene on metaphase spreads of the patient. We confirmed the presence of various BCOR exons on the inserts of the fosmids by STS typing (Figure 2b). Fosmid clone 2501D4 produced a signal on both X chromosomes (data not shown), whereas fosmid 1422H2 yielded a strong signal on one X chromosome and a weak signal on the second one (Figure 2c). Moreover, we observed only a single signal for fosmids 7601G5, 5351A7, and 9225G6 (Figure 2d and data not shown), suggesting that a larger deletion is present on one of the patient's X chromosome. We delineated the distal deletion breakpoint which seems to be present on fosmid clones 9478F10 and 2316B5 as both produced a strong and a weak signal on the patient's X chromosomes (data not shown). In contrast, fosmid clone 5335D1 showed two strong signals on both X chromosomes (data not shown), suggesting that this clone does not cover the deletion. We excluded the presence of the deletion on the X chromosomes of the parents of patient 3 by FISH (data not shown), indicating that it occurred apparently de novo. Taken together, our FISH data show the presence of a minimal deletion of approximately 60 kb on one X chromosome of patient 3 encompassing at least BCOR exons 2–15.
No BCOR mutation in patients with Lenz microphthalmia syndrome
The identification of a missense mutation in BCOR in one family resulting in a p.P85L change in all members affected by Lenz microphthalmia syndrome (MAA2)3 suggested that further mutations in this gene should be identified in male patients with MAA. We ascertained eight patients showing the characteristic clinical features of Lenz microphthalmia syndrome (patients 1–3) or a phenotype similar to this disorder (patients 4–8) (Table 2). We identified four sequence variations in BCOR (c.1260T>C, c.1692A>G, c.1791C>T, and c.4977-4G>T) that were also found in the SNP database or in controls, suggesting that they are not associated with the disease.
Discussion
We identified novel mutations in BCOR in three patients with OFCD syndrome and confirmed that BCOR is the causative gene for this disorder. The presence of two frameshift mutations and one large deletion encompassing almost the entire BCOR gene suggests that the causative mutations most likely lead to a functional null allele.3 Indeed, Ng et al3 already proposed that nonsense-mediated mRNA decay is the probable mechanism involved in preventing aberrant BCOR proteins to be expressed in vivo.7
In one patient with OFCD syndrome, we identified a submicroscopic deletion. The large deletion in one OFCD patient described by Ng et al3 encompasses at least exons 9–15 of BCOR covering a minimum of 20 kb, whereas the ∼60-kb deletion described in this report most likely spans exons 2–15. It is of interest to note that BCOR is located in a gene-poor region and is flanked distally by the STRAIT11499/MIG12 gene, located about 1.3 Mb distal to BCOR, whereas the ATP6AP2 gene lies about 500 kb proximal. Heterozygous microdeletions in females with X-linked dominant diseases with or without male lethality, such as incontinentia pigmenti (IP), oral-facial-digital syndrome type 1 (OFD1), and Rett syndrome, are not uncommon.8, 9, 10 Consequently, a careful mutation analysis including, for example, SNP haplotype analysis, multiplex amplification and probe hybridization (MAPH), multiplex ligation-dependent probe amplification (MLPA),11 and/or FISH with fosmid clones is required not to miss gross rearrangments undetectable by conventional cytogenetic analysis.
We did not identify a pathogenic mutation in BCOR in eight patients with Lenz microphthalmia or related phenotypes. Nonetheless, four sequence variations have been detected that do not seem to be associated with the disease. Only a single missense mutation has been described in BCOR in a large African-American family with six affected males exhibiting variable features of microphthalmia or anophthalmia, microcephaly, mental retardation, renal aplasia, cryptorchidism, and hypospadias (MAA2).3, 5 The amino-acid residue affected by the mutation, p.P85, is highly conserved in BCOR proteins of other species. However, a comparison of the clinical features of the patients described by Ng et al5 with those originally reported by Lenz6 showed that the latter ones presented with digital anomalies including double thumbs, clinodactyly, and hypoplasia of the terminal phalanges which are absent in the MAA2 patients. Anomalies of the fingers and toes have been reported for numerous patients with Lenz microphthalmia syndrome,4, 12, 13, 14, 15, 16, 17 suggesting that these represent important diagnostic criteria. Thus, the patients described by Ng et al5 show some overlapping features with Lenz microphthalmia syndrome, but the absence of digital anomalies with exception of extra flexion creases of fingers in one of them questions the initial diagnosis. It seems more likely that these patients show a unique phenotype comparable to, for example, Prieto syndrome that has been exclusively described in a single three-generation Spanish family.18 In conclusion, while the p.P85L change in BCOR might represent the pathogenic mutation in the family described by Ng et al,5 BCOR mutations appear not to be a frequent cause of Lenz microphthalmia syndrome, on the basis of our data. Based on the linkage data reported by Forrester et al,4 the candidate region for Lenz microphthalmia syndrome (MAA) is located in Xq27–q28 between markers DXS1232 and DXS8043 spanning a physical region of about 5 Mb. So far, the causative gene (which remains to be identified) also represents a suitable candidate for other patients with the clinical signs of Lenz microphthalmia syndrome. Further studies are necessary to identify the major gene causative for this condition.
References
Hedera P, Gorski JL : Oculo-facio-cardio-dental syndrome: skewed X chromosome inactivation in mother and daughter suggest X-linked dominant inheritance. Am J Med Genet 2003; 123A: 261–266.
Schulze BR, Horn D, Kobelt A, Tariverdian G, Stellzig A : Rare dental abnormalities seen in oculo-facio-cardio-dental (OFCD) syndrome: three new cases and review of nine patients. Am J Med Genet 1999; 82: 429–435.
Ng D, Thakker N, Corcoran CM et al: Oculofaciocardiodental and Lenz microphthalmia syndromes result from distinct classes of mutations in BCOR. Nat Genet 2004; 36: 411–416.
Forrester S, Kovach MJ, Reynolds NM, Urban R, Kimonis V : Manifestations in four males with and an obligate carrier of the Lenz microphthalmia syndrome. Am J Med Genet 2001; 98: 92–100.
Ng D, Hadley DW, Tifft CJ, Biesecker LG : Genetic heterogeneity of syndromic X-linked recessive microphthalmia-anophthalmia: is Lenz microphthalmia a single disorder? Am J Med Genet 2002; 110: 308–314.
Lenz W : Recessive, sex-limited microphthalmia with multiple abnormalities. Z Kinderheilkd 1955; 77: 384–390.
Baker KE, Parker R : Nonsense-mediated mRNA decay: terminating erroneous gene expression. Curr Opin Cell Biol 2004; 16: 293–299.
Aradhya S, Woffendin H, Jakins T et al: A recurrent deletion in the ubiquitously expressed NEMO (IKK-gamma) gene accounts for the vast majority of incontinentia pigmenti mutations. Hum Mol Genet 2001; 10: 2171–2179.
Laccone F, Junemann I, Whatley S et al: Large deletions of the MECP2 gene detected by gene dosage analysis in patients with Rett syndrome. Hum Mutat 2004; 23: 234–244.
Morisawa T, Yagi M, Surono A et al: Novel double-deletion mutations of the OFD1 gene creating multiple novel transcripts. Hum Genet 2004; 115: 97–103.
Sellner LN, Taylor GR : MLPA and MAPH: new techniques for detection of gene deletions. Hum Mutat 2004; 23: 413–419.
Baraitser M, Winter RM, Taylor DS : Lenz microphthalmia – a case report. Clin Genet 1982; 22: 99–101.
Herrmann J, Opitz JM : The Lenz microphthalmia syndrome. Birth Defects Orig Art Ser 1969; V: 138–143.
Glanz A, Forse A, Polomeno RC, Cole DE : Lenz microphthalmia: a malformation syndrome with variable expression of multiple congenital anomalies. Can J Ophthalmol 1983; 18: 41–44.
Antoniades K, Tzouvelekis G, Doudou A, Nanas C : A sporadic case of Lenz microphthalmia syndrome. Ann Ophthalmol 1993; 25: 342–345.
Temtamy SA, Ismail SI, Meguid NA : Lenz microphthalmia syndrome: three additional cases with rare associated anomalies. Genet Couns 2000; 11: 147–152.
Traboulsi EI, Lenz W, Gonzales-Ramos M, Siegel J, Macrae WG, Maumenee IH : The Lenz microphthalmia syndrome. Am J Ophthalmol 1988; 105: 40–45.
Prieto F, Badia L, Mulas F, Monfort A, Mora F : X-linked dysmorphic syndrome with mental retardation. Clin Genet 1987; 32: 326–334.
Acknowledgements
We thank Isabella Wimplinger for excellent help with FISH analysis. This work was supported by grants of the Deutsche Forschungsgemeinschaft (GRK336 and Wi 1140/6-1).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Horn, D., Chyrek, M., Kleier, S. et al. Novel mutations in BCOR in three patients with oculo-facio-cardio-dental syndrome, but none in Lenz microphthalmia syndrome. Eur J Hum Genet 13, 563–569 (2005). https://doi.org/10.1038/sj.ejhg.5201391
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.ejhg.5201391
Keywords
This article is cited by
-
CUGC for syndromic microphthalmia including next-generation sequencing-based approaches
European Journal of Human Genetics (2020)
-
Clinical presentation and genetic profiles of Chinese patients with velocardiofacial syndrome in a large referral centre
Journal of Genetics (2019)
-
Expanding the phenotype of the X-linked BCOR microphthalmia syndromes
Human Genetics (2019)
-
Ocular findings in a patient with oculofaciocardiodental (OFCD) syndrome and a novel BCOR pathogenic variant
International Ophthalmology (2018)
-
Recurrent BCOR internal tandem duplication and BCOR or BCL6 expression distinguish primitive myxoid mesenchymal tumor of infancy from congenital infantile fibrosarcoma
Modern Pathology (2017)
