
Abstract
Segmental aneuploidy usually has phenotypic consequences but unbalanced rearrangements without phenotypic consequences have also been reported. In particular, harmless deletions of G-dark bands 5p14 and 16q21 have each been found in more than one independent family. Here, we report two families that were ascertained at prenatal diagnosis and had similar overlapping deletions that removed most of the gene poor G-dark band 2p12. PCR mapping showed that the deletions had a minimum size of 6.1 and 6.9 Mb with at least 13 hemizygous loci including a cluster of six pancreatic islet-regenerating genes. These deletions had no apparent phenotypic consequences in eight family members. In contrast, a third family was ascertained through a child with Wilm's tumour; both the child and his mother had more proximal deletions, developmental delay and some dysmorphic features. The deletion had a minimum size of 5.7 Mb and extended into the gene-rich area of 2p11.2. These results are consistent with the idea that there may be segments of the genome that are consistently haplosufficient. The introduction of higher resolution methods of dosage analysis into diagnostic laboratories is already revealing more transmitted abnormalities of uncertain significance. As a result, published cases of transmitted imbalances have been collected as a guide to the possible significance of such findings in the future (see the ‘Chromosome Anomaly Collection’ at www.som.soton.ac.uk/research/geneticsdiv).
Similar content being viewed by others


Introduction
It can no longer be assumed that euchromatic imbalance at the cytogenetic level is automatically associated with mental and physical impairment. Exceptions to this rule have been found in families in which autosomal deletions or duplications have been directly transmitted from phenotypically normal parents to unaffected children. Couturier et al1 reported a clear example in 1985 when a deletion of 13q21 was found in a mother and son with normal phenotypes. The following year, Overhauser et al2 used molecular genetic methods to confirm a deletion of 5p14 in six phenotypically normal carriers from a three-generation pedigree. However, it was not until 14 years later that Hand et al3 confirmed these observations by finding a similar 5p14 deletion in a child referred for a peroxisomal disorder that was regarded as coincidental to the deletion.
A larger number of transmitted and de novo abnormalities of uncertain clinical significance are being detected with the increasing resolution of conventional cytogenetic analysis,4 molecular cytogenetic analysis5, 6, 7, 8 and the introduction of array technology into diagnostic laboratories.9 Transmitted imbalances are one of the few ways in which the significance of this new class of abnormalities can be determined. Here, we report the direct transmission of deletions of the proximal short arm of chromosome 2 from parents to children in two independent families without apparent phenotypic consequences and a third family in which the deletion is associated with a clinical phenotype. The two clinically normal families with 2p12 deletions suggest that at least 13 genes within band 2p12 are haplosufficient.
Material and methods
Probes
Family 1
Whole chromosome paint (wcp) for chromosome 2 was obtained from Cambio. YACs 850A4 (D2S2112), 747F5 (D2S2110) and 953D7 (D2S329) were mapped to 2p12 and provided by the YAC Screening Centre (Milan).
Family 2
The Multicolour banding (MCB) probe set for chromosome 2 was described by Weise et al10 and YAC 894F8 in 2p13.1–12.3,. YAC 953D7 in 2p12 and BAC 4C8 in 2p11.2 were used for conventional FISH.
Cytogenetic and molecular cytogenetic methods
Chromosome preparations were prepared using standard techniques after synchronisation with excess thymidine and release with deoxycytidine.11 Fluoresecence in situ hybridisation in families 1 and 2 was carried out using modifications of the method of Pinkel et al,12 the MCB method in family 2 as described by Weise et al10 and the high-resolution comparative genomic hybridisation (HR-CGH) in family 3 according to the method of Kirchhoff et al.5
Molecular genetic methods
The extent of the deleted material in families 1, 2 and 3 was determined by microsatellite analysis using fluorescent PCR. Physical locations were taken from the Ensembl database (www.ensembl.org) Version 19.34a1 (15th December, 2003). All the primer sequences and PCR conditions are available from the Genome Database (www.gdb.org). For family 1, the breakpoints were refined using replicate semiquantitative PCR. Each test was performed in duplicate with each microsatellite being coamplified with a control disomic sequence for the deletion carrier and a number of normal controls. Standard PCR conditions were used but the number of cycles was reduced to 20 to ensure linear amplification as described by Roberts and Thomas.13 All PCR products were analysed on an ABI automated sequencer (Applied Biosystems, Foster City, CA, USA).
The breakpoints in family 3 were refined using real-time quantitative PCR on an ABI Prism 7000 sequence detection system (Applied Biosystems, Foster City, CA, USA) according to the manufacturer's instructions. Primers were designed with Primer Express Software (Applied Biosystems, Foster City, CA, USA) and DNA sequence information was obtained from the public UCSC database (www.genome.ucsc.edu) (UCSC version hg16; NCBI build 34). An SYBR Green PCR master mix (Applied Biosystems, Foster City, CA, USA) was used for PCR according to the manufactureŕs instructions. The copy number was measured relative to GAPDH. Two normal individuals were included. Each assay was duplexed and evaluated by a comparative method validated by Applied Biosystems with the formula 2−ΔΔCt. Care was taken that as equal as possible amounts of reference and patient DNA were compared, illustrated by the GAPDH Ct value.
Results
Cytogenetics and molecular cytogenetics
Family 1
Nondisjunctional trisomy 18 was found in the proband (III. 1) (Figure 1). After amniocentesis in her next pregnancy at the age of 21 years, an interstitial deletion of chromosome 2 (del (2)(p11.2p13)) (Figure 2) was identified in the mother (II.3) and foetus (III.2). The same deletion was found after amniocentesis in her subsequent pregnancy (III.3). In addition, a paternal balanced pericentric inversion of chromosome 4 (inv(4)(p15.32q21.3)) was found in both pregnancies. The deletion was present in a total of five family members including a maternal aunt (II.5) and the maternal grandmother (I.2). These family members were phenotypically normal as were the children born from the two pregnancies at term. The mother was overweight from the age of 11 and the grandmother at the age of 74 years, suffers from severe obesity and hypertension.

Pedigree of family 1. The affected proband with trisomy 18 is fully shaded. Upper right quarter shading indicates individuals with a deletion of 2p, lower right quarter shading denotes chromosome 4 inversion carriers and ‘N’ indicates chromosomally normal family members.

Idiogram and partial karyotypes of the deletion in family 1. The deleted chromosome is on the right-hand side in each case and the deleted region arrowed.
In a lymphoblastoid cell line from the grandmother (I.2), wcp hybridised to both chromosomes 2 uniformly and to no other chromosome (data not shown). FISH with the proximal 2p12 YAC 953D7 (D2S329) confirmed a deletion of part of this band (Figure 3A), while the distal 2p12 YACs 850A4 (D2S2112) and 747F5 (D2S2110) were retained on both homologues. The karyotype of the grandmother was: 46,XX,del(2)(p11.2p12).ish del(2)(wcp2+, D2S2112+, D2S2110+, D2S329−). This lymphoblastoid cell line is available from the European Cell and Animal Culture Collection (ECACC) (ref. DD 0141).

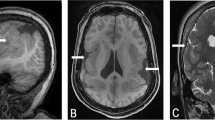
(A) The white arrow indicates the deletion of YAC 953D7 using FISH in family 1; (B, a) MCB in family 2 using probe set 2. One of the dark greenish pseudocolour bands indicated by the white arrows on the pseudocoloured chromosomes and fluorochrome profiles is absent from the deleted 2 and present on the normal 2; (B, b) Results of conventional FISH illustrating the deletion of YAC 953D7 in family 2. (C) HR-CGH in family 3 showing the 99.5% confidence intervals (light lines) and 99.5% dynamic standard reference intervals (dark lines). The deletion at chromosome 2p is detected where the two intervals do not overlap. Vertical lines represent the 0.5, 0.75, 1.0, 1.25 and 1.5 ratios. The deletion is shown as a red bar to the left of the chromosome idiogram. For technical reasons, the length of the bar is somewhat larger than the actual size of the deletion.
Family 2
A morphologically altered chromosome 2 was found in the foetus, his mother and her father after a woman of 38 years was referred for amniocentesis due to advanced maternal age. According to GTG banding, both breakpoints of the altered chromosome were thought to be in 2p12 (data not shown). The karyotype was refined to 46,XX or XY,del(2)(p11.2p12 or p12p12).ish del(2)(894F8+;953D7-;4C8+) after applying the region-specific 2p13.1–p12.3 YAC 894F8 (Figure 3B), the 2p12 YAC 953D7 and the 2p11.2 BAC 4C8, as well as the high-resolution MCB probe set for chromosome 2 (Figure 3B). The missing chromosomal material could not be detected elsewhere in the karyotype of any of the three family members studied using these FISH techniques. The foetus was born as a normal male and the mother and grandfather were of normal intelligence. Further family members were not available for cytogenetic studies.
Family 3
The proband was ascertained at 11 months of age when he was treated for Wilms tumour and it was noticed that he was physically and mentally retarded. His chromosomes were normal at the level of resolution available (Figure 4), but a diminution of proximal 2p was subsequently detected in the proband (dim(2p10p12)mat) and his mother (Figure 3C) after a series of patients with apparently normal karyotypes were referred to the Juliane Marie Centre for testing with HR-CGH. He walked at 20 months and, at 2 years and 4 months, all his milestones were delayed corresponding to a developmental age ranging between 11 and 21 months; language was the most severely affected at the equivalent of 11 months of age. He had problems concentrating but was cheerful and easy to communicate with. There was a suspicion of epilepsy. He had a relatively large head with frontal bossing, a flat face and low-set abnormally moulded ears. No hearing loss or visual problems were recorded.

Partial karyograms of chromosomes 2 from the proband in family 3. The deletion could not be confirmed using GTG-banding analysis.
He had a recurrence of Wilms localised to the right kidney, a nephrectomy and chemotherapy but is now generally well with no indication of a recurrence. He attends a special kindergarten and his development is progressing. At 4 years and 9 months, he has a developmental age of 2 years and 6 months. He still has problems concentrating, is easily diverted and has substantial problems with his pronunciation.
The mother has features similar to the proband. In particular, she has the same abnormally moulded ears, which were operated on for aures alatae, and severe pronunciation problems. She had difficulties in school and her expressive language problems make her difficult to understand. She has limited intellectual resources but takes care of her five children as a single parent.
Molecular mapping
A clear region of hemizygosity within flanking regions of heterozygosity was identified in all family members tested using 29 microsatellites across the 15.9 Mb of chromosome 2 between 72.4 Mb and 88.3 Mb from the 2p telomere. The grandmother was mapped in family 1, all three deletion carriers in family 2 and both deletion carriers in family 3. Replicate dosage PCR was needed to complete the molecular mapping in family 1 to distinguish between homozygosity and hemizygosity for a number of markers just within and outside the deletion. Families 1 and 2 had similar sized deletions with minimum sizes of 6.1 and 6.9 Mb and maximum sizes of 7.4 Mb (Figure 5). Both distal breakpoints were within the 0.5 Mb between D2S286 and D2S2114 and both proximal breakpoints within the 1.2 Mb between D2S2343 and D2S1396 (Figure 5).

Extent of each deletion with respect to the cytogenetic bands of proximal 2p and the physical location of the microsatellites tested. Deleted loci are shaded in black.
Family 3 had a more proximal deletion with a distal breakpoint within the 1.2 Mb between D2S2180 and D2S2343 and a proximal breakpoint within the 1.7 Mb between D2S1331 and D2S2216 (Figure 5). These estimates were further refined in family 3 using real-time quantitative PCR. Amplicons showing reduced copy number were positioned on chromosome 2 at 81212491 and 86900295 bases from the short arm telomere, giving a minimum size of 5.69 Mb. Amplicons showing normal copy number were positioned at 80792535 and 88444328 bases indicating a maximum size of 7.65 Mb. However, the most proximal amplicon showing normal copy number was at 88311785 using D2S2216 microsatellite typing and at 88444328 using real-time PCR. Thus, when these microsatellite and real-time PCR results are combined, the maximum size of the deletion is reduced to 7.52 Mb.
Discussion
The deletion in family 1 was originally reported in abstract.14 The apparently harmless deletions of 2p12 in eight individuals from families 1 and 2 contrast with the more proximal 2p11.2–2p12 deletion in family 3 and the relatively consistent phenotype in the four patients reviewed by Lacbawan et al.15 These four patients had overlapping deletions of 2p11–p13 or 2p11.2–p13, which extended into both the flanking G-light bands 2p11.2 and 2p13. Consistent findings included abnormal facies with a prominent or broad nasal bridge, an abnormally shaped nose and low set or abnormal ears. Myopia and sensorineural or conductive hearing loss were also found. The proband in family 3 had frontal bossing, abnormal low-set ears and developmental delay but no recorded visual or hearing loss. The mother had similar features and both the mother and child had language problems. We conclude that the phenotype in both of them is due to the 7.5 Mb deletion, which extends into gene-rich 2p11.2. This is analogous to the deletions of band 5p14; those restricted to G-dark 5p14 itself have no consistent phenotypic effect,2 while those that extend into the adjacent G-light band 5p13 are associated with mental retardation in multiple family members.16
G-dark bands are generally gene poor17 and the 8.3 Mb of 2p12 contain only 15 genes. The deletions in families 1 and 2 are a minimum of 6.1 and 6.9 Mb in size, respectively, and contain at least 13 hemizygous genes (Table 1). In addition, at least part of the tachykinin 1 receptor locus (TACR1) is deleted in family 1. Six of these 13 belong to the precursor islet of langerhans-regenerating family (REG). Underexpression of these genes has been associated with chronic calcifying pancreatitis and overexpression with diabetes. This raises the possibility of a ‘cryptic’ pancreatic phenotype, which might only be expressed as a result of damage or infection of the pancreas. The corollary might be protection against the heritable causes of diabetes. Alternatively, haploidy of this cluster of closely related genes, which is thought to have arisen by duplication, may be more easily tolerated than haploidy of a series of unrelated single loci. The majority of the other genes involved also have related loci on other chromosomes (Table 1). Our results imply that either all the deleted loci are haplosufficient or that the cumulative effect of these oligogenetic imbalances is insufficient to disturb normal development. It has also been suggested that 2p12 is haplolethal because deletion of this band has not been previously recorded in the Human Cytogenetics Database.18 In contrast, our results suggest that the deletions in families 1 and 2 are rarely ascertained because any phenotypic consequences would be unlikely to come to medical attention.
Other families in which deletions, duplications or unbalanced translocations have been transmitted from an unaffected parent to an unaffected or affected child are listed in Tables 2 and 3, respectively. Marker chromosomes have been specifically excluded as they have been extensively reviewed elsewhere. In Table 2, the majority of families (11/15) were ascertained at prenatal diagnosis or for other reasons that are likely to have been coincidental. The chromosomal imbalances have been confirmed by a complementary technique in 12/15 families. Bortotto et al19 proposed that the predominantly maternal transmission could be associated with imprinting, but none of the regions in Table 2 is thought to contain imprinted loci. Incomplete ascertainment of these families is a more likely explanation. These imbalances are not thought to have been responsible for any phenotypic effects among the 43 carriers.
In Table 3, the majority of families (17/21) were ascertained through the phenotype of an affected proband, but the parent and other relatives with the same imbalance were unaffected. The imbalance was confirmed in 14/21 families. These abnormalities are analogous to those in Table 2 wherever the phenotype in the proband is likely to have been coincidental as in the del(5) of Hand et al,3 the del(11) of Li et al,20 the dup(1) of Bortotto et al,19 the dup(1) of Chan et al,21 the dup(8)s of Harada et al22 and the der(15) of Guichaoua et al.23 Imprinting explains the contrast between the phenotypes in parents and children with duplications of 15q11.2–q13,24 6q2425 and possibly 14q24.3–q31.26 Re-examination of the parents in the case of Browne et al24 suggests that paternal duplications of 15q11.2-q13 can have phenotypic consequences, but much milder than those associated with maternal duplications of the same segment.27 The lack of a ‘second hit’ is likely to explain the lack of retinoblastoma in the mother of a child with the 13q14 deletion.28 Nevertheless, over half the 56 carriers in this category were phenotypically unaffected.
To the best of our knowledge, only the duplications of 8p23.222 and the deletions of 5p14,2, 29 16q213 and 2p12 (present report) have been transmitted without consistent phenotypic consequences in more than one independent family. We suspect that many more families exist than are reported in the literature and that segmental haplosufficency may be more common at the cytogenetic and subcytogenetic level, especially where gene density is low. These results have implications for microarrays and other high-resolution means of dosage analysis as not all the abnormalities detected will be causal. In addition, arrays in which probe density reflects gene density may be a better way of detecting clinically significant cases than arrays in which probes are spaced at regular intervals. A collection of other published examples of apparent haplosufficiency or chromosomal nonpenetrance (the ‘Chromosome Anomaly Collection’) can be viewed at www.som.soton.ac.uk/research/geneticsdiv/.
References
Couturier J, Morichon-Delvallez N, Dutrillaux B : Deletion of band 13q21 is compatible with normal phenotype. Hum Genet 1985; 70: 87–91.
Overhauser J, Golbus MS, Schonberg SA, Wasmuth JJ : Molecular analysis of an unbalanced deletion of the short arm of chromosome 5 that produces no phenotype. Am J Hum Genet 1986; 39: 1–10.
Hand JL, Michels VV, Marinello MJ, Ketterling RP, Jalal SM : Inherited interstitial deletion of chromosomes 5p and 16q without apparent phenotypic effect: further confirmation. Prenat Diagn 2000; 20: 144–148.
Joyce CA, Dennis NR, Cooper S, Browne CE : Subtelomeric rearrangements: results from a study of selected and unselected probands with idiopathic mental retardation and control individuals by using high-resolution G-banding and FISH. Hum Genet 2001; 109: 440–451.
Kirchhoff M, Rose H, Lundsteen C : High resolution comparative genomic hybridisation in clinical cytogenetics. J Med Genet. 2001; 38: 740–744.
Martin CL, Waggoner DJ, Wong A et al: ‘Molecular rulers’ for calibrating phenotypic effects of telomere imbalance. J Med Genet. 2002; 39: 734–740.
Liehr T, Schreyer I, Neumann A et al: Two more possible pitfalls of rapid prenatal diagnostics using interphase nuclei. Prenat Diagn 2002; 22: 497–499.
Starke H, Nietzel A, Weise A et al: Small supernumerary marker chromosomes (SMCs): genotype–phenotype correlation and classification. Hum Genet 2003; 114: 51–67.
Vissers LE, de Vries BB, Osoegawa K et al: Array-based comparative genomic hybridization for the genomewide detection of submicroscopic chromosomal abnormalities. Am J Hum Genet 2003; 73: 1261–1270.
Weise A, Starke H, Heller A et al: Chromosome 2 aberrations in clinical cases characterised by high resolution multicolour banding and region specific FISH probes. J Med Genet 2002; 39: 434–439.
Wheater RF, Roberts SH : An improved lymphocyte culture technique: deoxycytidine release of a thymidine block and use of a constant humidity chamber for slide making. J Med Genet 1987; 24: 113–115.
Pinkel D, Landegent J, Collins C et al: Fluorescence in situ hybridization with human chromosome-specific libraries: detection of trisomy 21 and translocations of chromosome 4. Proc Natl Acad Sci USA. 1988; 85: 9138–9142.
Roberts SE, Thomas NS : A quantitative polymerase chain reaction method for determining copy number within the Prader–Willi/Angelman syndrome critical region. Clin Genet 2003; 64: 76–78.
Lambert R, Collinson MN : Familial microdeletion of chromosome 2 without apparent phenotypic effect. J Med Genet 1991; 28: 62.
Lacbawan FL, White BJ, Anguiano A et al: Rare interstitial deletion (2)(p11.2p13) in a child with pericentric inversion (2)(p11.2q13) of paternal origin. Am J Med Genet 1999; 87: 139–142.
Keppen LD, Gollin SM, Edwards D, Sawyer J, Wilson W, Overhauser J : Clinical phenotype and molecular analysis of a three generation family with an interstitial deletion of the short arm of chromosome 5. Am J Med Genet 1992; 44: 356–360.
Saccone S, Federico C, Solovei I, Croquette M-F, Valle GD, Bernardi G : Identification of the gene-richest bands in human prometaphase chromosomes. Chromosome Res 1999; 7: 379–386.
Brewer C, Holloway S, Zawalnyski P, Schinzel A, FitzPatrick D : A chromosomal duplication map of malfomations: regions of suspected haplo- and triplolethality and tolerance of segmental aneuploidy in humans. Am J Hum Genet 1996; 4: 1702–1708.
Bortotto L, Piovan E, Furlan R, Rivera H, Zuffardi O : Chromosome imbalance, normal phenotype, and imprinting. J Med Genet 1990; 27: 582–587.
Li L, Moore P, Ngo C et al: Identification of a haplosufficient 3.6-Mb region in human chromosome 11q14.3 → q21. Cytogenet Genome Res 2002; 97: 158–162.
Chan NPH, Ng MHL : Hereditary duplication of proximal chromosome 1q (q11q22) in a patient with T lymphoblastic lymphoma/leukaemia: a family study using G-banding and comparative genomic hybridisation. J Med Genet 2002; 39: e79.
Harada N, Takano J, Kondoh et al: Duplication of 8p23.2: a benign cytogenetic variant? Am J Med Genet 2002; 111: 285–288.
Guichaoua MR, Speed RM, Luciani JM, Delafontaine D, Chandley AC : Infertility in human males with autosomal translocations. 2. Meiotic studies in 3 reciprocal rearrangements, one showing tertiary monosomy in a 45-chromosome individual and his father. Cytogenet Cell Genet 1992; 60: 96–101.
Browne CE, Dennis NR, Maher E et al: Inherited interstitial duplications of proximal 15q: genotype-phenotype correlations. Am J Hum Genet 1997; 61: 1342–1352.
Temple IK, Gardner RJ, Robinson DO et al: Further evidence for an imprinted gene for neonatal diabetes localised to chromosome 6q22–q23. Hum Mol Genet 1996; 5: 1117–1123.
Robin NH, Harari-Shacham A, Schwartz S, Wolff DJ : Duplication 14(q24.3q31) in a father and daughter: delineation of a possible imprinted region. Am J Med Genet 1997; 71: 361–365.
Bolton PF, Dennis NR, Browne CE et al: The phenotypic manifestations of interstitial duplications of proximal 15q with special reference to the autistic spectrum disorders. Am J Med Genet 2001; 105: 675–685.
Cowell JK, Rutland R, Hungerford J, Jay M : Deletion of chromosome region 13q14 is transmissible and does not always predispose to retinoblastoma. Hum Genet 1988; 80: 43–45.
Johnson EI, Marinescu RC, Punnett HH, Tenenholz B, Overhauser J : 5p14 deletion associated with microcephaly and seizures. J Med Genet; 2000; 37: 125–127.
Sumption ND, Barber JCK : Transmitted deletion of 2q13 to 2q14.1 causes no phenotypic abnormalities. J Med Genet 2001; 38: 125–126.
Knight LA, Yong MH, Tan M, Ng ISL : Del(3)(p25.3) without phenotypic effect. J Med Genet 1995; 32: 994–995.
Reddy KS : A paternally inherited terminal deletion, del(8)(p23.1)pat, detected prenatally in an amniotic fluid sample: a review of deletion 8p23.1 cases. Prenat Diagn 1999; 19: 868–872.
Batanian JR, Morris K, Ma E, Huang Y, McComb J : Familial deletion of (8)(q24.13q24.22) associated with a normal phenotype. Clin Genet 2001; 60: 371–373.
Barber JCK, Mahl H, Portch J, Crawfurd MD : Interstitial deletions without phenotypic effect: prenatal diagnosis of a new family and brief review. Prenat Diagn 1991; 11: 411–416.
Zaslav AL, Blumenthal D, Fox JE, Thomson KA, Segraves R, Weinstein ME : A rare inherited euchromatic heteromorphism on chromosome. 1. Prenat Diagn 1993; 13: 569–573.
Engelen JJM, Moog U, Evers JLH, Dassen H, Albrechts JCM, Hamers AJH : Duplication of chromosome region 8p23.1 → p23.3: a benign variant? Am J Med Genet 2000; 91: 18–21.
Wolff DJ, Raffel LJ, Ferre MM, Schwartz S : Prenatal ascertainment of an inherited dup(18p) associated with an apparently normal phenotype. Am J Med Genet 1991; 41: 319–321.
Borgaonkar DS, Bias WB, Chase GA et al: Identification of a C6/G21 translocation chromosome by the Q–m and Giemsa banding techniques in a patient with Down's syndrome, with possible assignment of Gm locus. Clin Genet 1973; 4: 53–57.
Mascarello JT, Hubbard V : Routine use of methods for improved G-band resolution in a population of patients with malformations and developmental delay. Am J Med Genet 1991; 38: 37–42.
Maltby EL, Barnes ICS, Bennett CP : Duplication involving band 4q32 with minimal clinical effect. Am J Med Genet 1999; 83: 431.
Li S-Y, Gibson LH, Gomez K, Pober BR, Yang-Feng TL : Familial dup (5)(q15q21) associated with normal and abnormal phenotypes. Am J Med Genet 1998; 75: 75–77.
Cook Jr EH, Lindgren V, Leventhal BL et al: Autism or atypical autism in maternally but not paternally derived proximal 15q duplication. Am J Hum Genet 1997; 60: 928–934.
Schroer RJ, Phelan MC, Michaelis RC et al: Autism and maternally derived aberrations of chromosome 15q. Am J Med Genet 1998; 76: 327–336.
Verma RS, Kleyman SM, Conte RA : Variant euchromatic band within 16q12.1. Clin Genet 1997; 52: 446–447.
Fu W-N, Borgaonkar DS, Ladewig PP et al: Structural aberrations of the long arm of chromosome no. 22. Clin Genet 1976; 10: 329–336.
Pfeiffer RA, Kessel EK, Soer K-H : Partial trisomies of chromosome 21 in man. Two new observations due to translocations 19;21 and 4;21. Clin Genet 1977; 11: 207–213.
Acknowledgements
We are grateful to the YAC Screening Centre for providing YACs, PACs and cosmids and particularly grateful to Sarah Beal for preparing probes for FISH. We are also indebted to the patients who contributed additional samples for research. The image enhancement equipment used for parts of this work was provided by the Wellcome Trust and Trust Funds of Salisbury NHS Health Care Trust. TL was supported in part by Dr Robert Pfleger-Stiftung.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Barber, J., Thomas, N., Collinson, M. et al. Segmental haplosufficiency: transmitted deletions of 2p12 include a pancreatic regeneration gene cluster and have no apparent phenotypic consequences. Eur J Hum Genet 13, 283–291 (2005). https://doi.org/10.1038/sj.ejhg.5201267
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.ejhg.5201267
Keywords
This article is cited by
-
A recurrent deletion syndrome at chromosome bands 2p11.2-2p12 flanked by segmental duplications at the breakpoints and including REEP1
European Journal of Human Genetics (2015)
-
Chromosome abnormalities without phenotypic consequences
Journal of Applied Genetics (2007)
