
Abstract
The combination of multiple exostoses (EXT) and enlarged parietal foramina (foramina parietalia permagna, FPP) represent the main features of the proximal 11p deletion syndrome (P11pDS), a contiguous gene syndrome (MIM 601224) caused by an interstitial deletion on the short arm of chromosome 11. Here we present clinical aspects of two new P11pDS patients and the clinical follow-up of one patient reported in the original paper describing this syndrome. Recognised clinical signs include EXT, FPP, mental retardation, facial asymmetry, asymmetric calcification of coronary sutures, defective vision (severe myopia, nystagmus, strabismus), skeletal anomalies (small hands and feet, tapering fingers), heart defect, and anal stenosis. In addition fluorescence in situ hybridisation and molecular analysis were performed to gain further insight in potential candidate genes involved in P11pDS.
Similar content being viewed by others


Introduction
Previously we described a contiguous gene syndrome due to deletions of the proximal short arm of chromosome 11 in eight patients from four families.1 Initially this syndrome was referred to as DEFECT 11 syndrome,1 but as this was considered potentially discriminating or offending to the patients and family members the accepted term today is proximal 11p deletion syndrome (P11pDS).2 Alternatively, Potocki–Shaffer syndrome is also used. In the original patients co-occurence of multiple exostoses (EXT) and foramina parietalia permagna (FPP) were observed, although the latter sign shows reduced penetrance. Additional features observed in a subset of patients included facial dysmorphism and mental retardation (MR). More detailed clinical analysis in recent papers have added additional characteristics of this syndrome, including complex malformations of the brain.3,4
All patients have cytogenetic and/or molecular deletions of chromosome 11p11–p13 located in a ∼20 cM region between the centromere and D11S914, with the severity of the clinical spectrum being related to the extent of the deletion.1 Since the original report, review of the literature and additional studies have identified additional patients, but P11pDS remains very rare with less than 20 well described patients.1,3,4,5,6,7,8,9,10
Today the genes responsible for the two main characteristics of P11pDS have been identified. The deletion of the EXT2 gene is responsible for the development of multiple exostoses,6 with EXT2 encoding a glycosyltransferase involved in heparan sulfate biosynthesis.11 Inactivating mutations in EXT2 also cause multiple exostoses in nonsyndromic patients suffering from isolated hereditary multiple exostoses (HME).6,12 The characteristic skull ossification defect (FPP) in P11pDS is the result of the deletion or inactivation of the ALX4 transcription factor, which is located close to and proximal of the EXT2 gene.13,14 The role of other genes located within the respective deletion interval of the various patients remains to be elucidated.
Here we present two additional P11pDS patients and provide a clinical follow-up of one previously described patient. Molecular analyses were performed to gain more insight in the position and possible role of further candidate genes for the observed clinical signs and symptoms.
Materials and methods
Cytogenetic analysis and fluorescence in situ hybridisation (FISH)
All patients were investigated by conventional karyotyping of cultured lymphocytes from peripheral blood using GTG banding at a 550+ band level prior to or in this study.
The FISH studies were performed using metaphase spreads from cultured lymphocytes. A total of five different FISH probes (AN-G1, WT1, DO694, 1/IB1, and P5030) were analysed using a protocol described elsewhere.15 The probes map on chromosome 11 from p13 to the centromere in the given order. P1-clone AN-G1 and cosmid clone WT1 (kindly provided by A Poustka and M Dressler, Deutsches Krebsforschungszentrum, Heidelberg) represent segments of PAX6, the gene for aniridia (currently located at 31.85 Mb on 11p13), and WT1, the gene for Wilms tumor (at approximately 32.45 Mb on 11p13), respectively. P1-clone DO694 contains the 3′-end of the EXT2 gene (44.16 Mb on 11p11.2) and locus D11S2095 (44.30 Mb on 11p11.2).6 BAC clone 1/IB1 includes the complete IB1 gene, alias JIP1 or MAPK8IP1 (45.94 Mb on 11p11.2).16 The digoxigenin-labelled probe P5030 detecting the D11Z1 alphoid DNA (approximately 53.20–58.40 Mb on 11p11.11–q11) (Appligene-Oncor, Illkirch, France) was used to facilitate the identification of chromosomes.11 Microscopy was performed with Axiophot™ epifluorescence microscopes (Carl Zeiss, Jena, Germany) and the ISISTM digital imaging system (MetaSystems, Altlussheim, Germany). At least 10 metaphases with clear hybridisation signals were scored per probe.
Microsatellite analysis
Patients 1 and 2 and their parents were investigated by PCR analysis of 10 different polymorphic microsatellite markers of the pericentromeric region of chromosome 11: D11S935 (36.06 Mb on 11p13, 49.60 cM), D11S905 (41.01 Mb on 11p12, 55.70 cM), D11S1355 (42.96 Mb on 11p12, 58.10 cM), D11S1393 (44.03 Mb on 11p11.2), D11S903 (44.20 Mb on 11p11.2, within EXT2, 59.50 cM), D11S2095 (44.30 Mb on 11p11.2), D11S554 (44.96 Mb on 11p11.2), D11S1361 (44.98 Mb on 11p11.2, 61.30 cM), D11S1319 (62.50 cM), D11S1344 (46.19 Mb on 11p11.2, 62.50 cM), and D11S1326 (49.36 Mb on 11p11.12, 62.50 cM) (succession from 11p13 to 11p11), using standard PCR reactions. The determination of the deletion breakpoints was based on the presence or absence of parental alleles, not on band signal density. Patient 3 has been tested before using similar methods and markers, as described elsewhere.1
Electronic-database information
Accession numbers and URLs for data in this article are as follows: The genetics location database (http://cedar.genetics.soton.ac.uk/) and NCBI genome database (http://www.ncbi.nlm.nih.gov/genome) for genetic and physical positions of loci on chromosome.11
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim for the P11pDS contiguous gene syndrome [MIM 601224].
Results
Clinical analysis
Patient 1
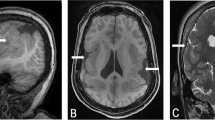
This female patient (Figure 1a, b) was born in 1977 to healthy German parents. Pre- and perinatal history was normal with birth weight 3000 g (P50) and length 52 cm (P90). However, hypotonia and developmental delay became soon evident. At 2.5 years multiple exostoses were diagnosed (Figure 1c). She sat without support at age 3 years and walked at age 7 years. Painful exostoses impairing articular function were removed by surgery. She had her menarche at age 11 years and regular bleedings. At 15 years she was referred to one of us (PM) who noted left facial asymmetry, high forehead, brachycephaly, large ears prominent high chin and high myopia at −10 dpt. In addition a small flat nose, and narrow maxilla were observed. There was severe MR with extreme delay of speech development. Following a seizure at age 17 years she was seen again. On physical examination of her skull parietal bone defects measuring 1–1.5 cm in size were noticed and subsequently the P11pDS was diagnosed. Cranial CT (Figure 1d) showed asymmetric hyperplasia of calvarial bones, a thickened coronary suture on the right and bilateral parietal foramina. The cerebellar vermis and hemispheres were markedly hypoplastic and the posterior horn of the right lateral ventricle was moderately enlarged. At her last visit at age 21 years her height was measured 160 cm (25<P<50) and her weight was 70 kg (P90). Occipital-frontal circumference (OFW) measured 54 cm (25<P<50). At age 25 years she is clearly obese with a weight of 88 kg.

Patient 1. Facial pictures at age 7 years (a) and 15 years (b) illustrating facial asymmetry, narrow pointed nose, high forehead, and large ears. (c) X-ray demonstrating exostoses on the radius; (d) Cranial CT showing parietal foramina. (e) Deletion of bands 11p11.2–p11.12 shown by karyotyping and (f) by FISH analysis with EXT2 probe D0694.
Patient 2
This girl, born in 1996, is the second child of young nonrelated parents of Albanian extraction. Pregnancy and birth at term were uncomplicated. Her birth weight and length were 3280 g (P90) and 51 cm (P75), respectively. By the age of 2 months anal atresia with anal fistula and two small muscular ventricular septal defects were observed. X-ray investigations (arm, hand, and vertebral column) did not show any abnormalities. At age 4 months muscular hypotonia became apparent as well as nystagmus. Length and weight were within normal limits while her head circumference measured 40.1 cm (P25). The occiput appeared flat and there was temporal narrowing, upslant of palpebral fissures, epicanthus, alternating convergent strabismus, horizontal nystagmus, and somewhat large ears (Figure 2a, b). A 2/6 systolic heart murmur, ventrally placed anus, small hands, and tapering fingers were observed. At age 2 years and 10 months, her height was 93.2 cm (25<P<50) and weight 16.5 kg (90<P<97). OFC measured 46.5 cm (<P3). She showed prominent glabella, full cheeks, narrow nose, hypoplastic alar wings, prominent chin, small teeth. and diastemata between maxillar and manibular incisivi. Her developmental milestones were markedly retarded with beginning to babble and sitting by age 2 years, standing unsupported at 6 years and walking by the age of 6½years. On re-examination at age 7 years and 2 months, she was 120 cm tall (25<P<50) and her weight was 23 kg (P50). Head circumference was 48.4 cm (<P3). She showed brachycephaly, a medially prominent forehead, narrow pointed nose, large ears, and diastemata. Both forearms showed radial bowing with restricted supination and extension in the right elbow. At both knees exostoses were palpable in the distal portion of the femora and proximal portion of the tibiae (Figure 2c). There was some restriction of extension in the left knee joint. Toes II/III showed syndactyly. She showed marked muscular hypotonia and slow muscular responses. She had profound MR with absent eye contact and produced chains of two syllables, but no meaningful words.

Patient 2. Facial pictures at (a) age 14 months and (b) 7 years showing a medially prominent forehead, narrow pointed nose, downturned mouth, large ears, and hypotelorism. (c) X-ray showing EXT in the distal portion of the femora and the proximal portion of the tibiae. (d) Karyotype depicting the 11p11.2–p11.12 deletion. (e) FISH using the IB1 DNA probe, note deletion on one chromosome 11 homolog.
Patient 3
This male patient (Figure 3) has been described previously 1,17. Here we report the clinical follow-up. At 14 years he was vital and lived with his mother. Height was approximately 143 cm (<P3), weight 25 kg (<P5), and head circumference 50 cm (<P3). He showed facial dysmorphism, turribrachycephaly, bilateral parietal foramina being 4 cm in diameter, strabismus, frequent fits of nystagmus, severe myopia (−7 to −10 dpt), numerous large painful exostoses and small hands and feet with tapering fingers. He had seizures since the age of 10 years. The psychomotor retardation was profound with a most severe attention deficit and autistic behaviour. He never walked alone, could not sit unsupported and is now wheelchair-bound. He could neither eat nor drink by himself and was averbal, even without comprehending his name. He had open eyes but showed no noticeable reaction to visual or acoustic stimuli.

Patient 3 at the age of 14 years showing, turribrachycephaly, facial asymmetry with antimongoloid eye slants and downturned mouth corners, high forehead, and narrow nose.
The major P11pDS features observed in the three patients are summarised in Table 1.
Cytogenetic and FISH Analysis
Patient 1
Initially the karyotype of patient 1 was regarded as normal. After clinically diagnosing the P11pDS, cytogenetic re-examination at 550 bands (GTG) revealed the deletion of bands p11.2–p11.12 on chromosome 11 (Figure 1e). FISH analysis showed the deletion at EXT2 and IB1 (Figure 1f). Parental karyotypes were normal. Her final karyotype read 46,XX,del(11)(p11.2p11.12).ish del(11)(p11.2p11.2)(PAX6+,WT1+,EXT2−,IB1−,D11Z1+) de novo.
Patient 2
Standard karyotyping at 550 bands resolution (GTG) revealed the deletion of bands p11.2–p11.12 on chromosome 11 (Figure 2d), without the P11pDS suspected on clinical grounds. FISH analysis shows the deletion at EXT2 and IB1 (Figure 2e). Parental karyotypes were normal. The final karyotype read 46,XX,del(11)(p11.2p11.12).ish del(11)(p11.2p11.2)(WT1+,EXT2−,IB1−,D11Z1+) de novo.
Patient 3
Cytogenetic analysis of the patient and his parents had been performed previously 1. FISH studies indicated the deletion at EXT2 and IB1. The combined karyotype was 46,XY,del(11)(p13p11.2).ish del(11)(p11.2p11.2)(PAX6+,WT1+,EXT2−,IB1−,D11Z1+) de novo.
Microsatellite analysis
Results are shown in Figure 4

Summary of the molecular analyses in published P11pDS patients. At the left, genetic markers and genes are listed with their physical position in megabases (Mb) from pter. At the right, the 11p deletions (represented by black boxes) of patients described in this study and previously published deletions in P11pDS patients are compared. Regions containing the deletion breakpoints are represented by white boxes. References for each patient are listed at the top. A phenotypic map showing the position of genes/candidate regions for multiple EXT, enlarged foramina (FPP), MR and hypertrophic cardiomyopathy (CMH) is included.
Patient 1
Results comprised biparental alleles at markers D11S935 (ND, not deleted), D11S905 (ND), and D11S1355 (ND). Only a maternal allele was observed for markers D11S1393 (DEL, deleted), D11S554 (DEL), and D11S1344 (DEL). No informative result were obtained for D11S903 (NI, not informative), D11S2095 (NI), and D11S1326 (NI). This corresponds to a paternal deletion encompassing a maximum of 10.2 Mb between D11S1355 (located at 42.96 Mb on 11p11.2) and the pericentric alphoid DNA (beginning at 53.20 Mb on 11p11.11).
Patient 2
A paternal deletion was observed for D11S1355 and D11S554 while D11S935, D11S905, and D11S1326 were not deleted. The remaining markers were not informative. This corresponds to a deletion with a maximum size of 8.35 Mb between D11S905 (located at 41.01 Mb on 11p11.2) and D11S1326 (located at 49.36 Mb on 11p11.12).
Patient 3
Molecular analysis was previously performed and shows a maximal deletion of approximately 17 Mb between D11S935 (36.06 Mb) and the centromere on the paternal chromosome.1
Discussion
P11pDS is a rare syndrome with less than 20 patients described in the literature today. We now report clinical, cytogenetic, FISH, and/or molecular findings in three nonrelated patients with the p11pDS, including two previously undescribed patients.
The two hallmarks of P11pDS are EXT and FPP. However, the age of manifestation of EXT is variable and FPP are a trait with variable penetrance.1 Therefore, the absence of EXT and FPP in patient 2 was not necessarily contradictory with a diagnosis of P11pDS at the time the 11p deletion was identified, as the absence of multiple exostoses could be attributed to her young age at the time of diagnosis (34 months) and the absence of FPP could be explained by reduced penetrance. Clinical follow-up at the age of 7 years indeed showed the occurrence of multiple exostoses.
Besides EXT and FPP a great variation in additional signs and symptoms have been described. Among these additional characteristics are structural brain abnormalities that have been added recently to the clinical spectrum after detailed clinical analysis of patients by CT and MRI. Also the only patient from this study for whom MRI data was available (patient 1) showed hypoplasia of the vermis and cerebellar hemispheres, similar to the cases described by Wuyts et al3 and Yamamoto et al.4 This suggests that these brain malformations may be under-recognised in previous reports in which no CT or MRI was performed and they may represent a more common feature of P11pDS.
P11pDS patients demonstrate striking differences in intelligence, a subset having normal or near-normal intelligence and another subset (including patients 1–3) showing severe to profound MR, strongly suggesting the presence of a major MR gene on proximal 11p. Most mentally retarded patients harbour deletions with more distal breakpoints, while the nonretarded individuals mostly harbour rather smaller deletions around the EXT2 gene. Review of the literature defines two candidate regions for the gene(s) causing profound MR in P11pDS: a distal region of approximately 3.2 Mb between D11S905 (41.01 Mb)3,4 and the EXT2 intragenic marker D11S903 (44.20 Mb) and a proximal 1.2 Mb region between D11S554 (44.96 Mb) and D11S1344 (46.19 Mb).1,3,4 The patients described here show profound or severe MR and their respective deletions include the proximal MR region. At the distal side, however, the borders of the deletion extend much less distal compared to previous described P11pDS patients with severe MR and they reduce the distal MR candidate region to approximately 1.2 Mb between D11S1355 (42.96 Mb) and D11S903 (44.20 Mb).
In contrast to EXT and FPP, the craniofacial abnormalities in PD11pDS are less well documented. Mild facial dysmorphism with brachycephaly and high forehead has been described in several patients,1,4,8,10,17 but clear-cut craniofacial dysmorphism was only reported in a single patient (patient 3 of this report). In this study, we observed facial asymmetry, hypotelorism, narrow maxilla and mandible, hyperplasia of calvarial bones, and thickened coronary suture in patient,1 compatible with a mild craniosynostosis syndrome.
Myopia is a common condition with heterogeneous aetiology as it is found in monogenic disorders of dominant (e.g. Stickler syndrome, Marfan syndrome) and recessive (e.g. Cohen syndrome, homocystinuria) inheritance. In patient 1 high myopia is combined with eye movement disorders strabismus and nystagmus, which are frequently observed in patients with MR, including p11p11DS patients.1,8,18 Since myopia can progress with advancing years and may even give rise to retinal detachment and, ultimately, blindness, periodical ophthalmologic check-ups are recommended in individuals with P11pDS and MR.
The 11p11 region harbours several genes involved in the development of organs and structures affected in P11pDS patients, but with exception of EXT2 and ALX4, a clear correlation between heterozygous deletion of these genes and observed P11pDS phenotype has not yet been deducted. Mutations in some of these genes, such IB1, MYBPC3 or PEX16 have been associated with a disease phenotype. The human islet-brain-1 (IB1) or MAPK8IP1 gene is a DNA-binding transactivator of the glucose transporter GLUT2 and shows high expression in pancreas, brain, testis, retina, and prostate.16 A single missense mutation in IB1 was reported to be associated with in a family with type II diabetes.19 The effect of heterozygous inactivation of this gene is yet unknown, but in vitro and animal studies have indicated an important role of this gene in the hippocampus20 making it a serious candidate to contribute to the MR observed in patients with deletions encompassing this gene. The deletion in patients 1 and 3 includes the MYBPC3 gene (47.39 Mb), encoding the cardiac myosin binding protein C. This protein is thought to regulate cardiac contractility by binding to myosin heavy chain and the cytoskeleton protein titin and truncating mutations in the MYBPC3 are known to cause hypertrophic cardiomyopathy.21,22 At present none of the patients described in this study shows cardiac abnormalities. It should be noted, however, that hypertrophic cardiomyopathy caused by MYBPC3 mutations is characterised by reduced penetrance until the fifth decade, so the patients with MYBPC3 deletion may still be at risk for developing cardiac problems at later age. The PEX16 gene (45.96 Mb) encodes the peroxisomal protein peroxin.16 Homozygous inactivating PEX16 mutations have been detected in patients with Zellweger syndrome of complementation group D,23 but heterozygous carriers are normally asymptomatic and heterozygous deletion of PEX16 is therefore not likely to contribute to the clinical P11pDS spectrum.
NEGF2, also known as midkine (MDK), is a member of a highly conserved family of developmental genes in humans. The gene product exhibits neurite outgrowth-promoting activity and may play a role in nervous system development and/or maintenance. Although positioned at 46.4 Mb and thus located outside the proximal MR critical region if previously published patients are considered, the NEGF2 gene was found deleted in the three patients, all with severe or profound MR, presented here. Therefore, NEGF2 may contribute to the MR spectrum in these patients. The P11pDS region also harbours the gene for lysosomal acid phosphatase (ACP2), a tartrate sensitive enzyme with ubiquitous expression. Animal studies have shown that a proportion of Acp −/− KO mice develop generalised seizures24 making ACP2 an attractive candidate gene for the epilepsy that has been observed in P11pDS patients.
In conclusion, we have provided detailed clinical and molecular data that may lead to a better understanding of the underlying biological processes causing the P11pDS phenotype. However, further characterisation of P11pDS patients is required for identifying all the genes contributing to this syndrome.
References
Bartsch O, Wuyts W, Van Hul W et al: Delineation of a contiguous gene syndrome with multiple exostoses, enlarged parietal foramina, craniofacial dysostosis and mental retardation, caused by deletions on the short arm of chromosome 11. Am J Hum Genet 1996; 58: 734–742.
Wuyts W, Bartsch O, Wilkie AO, Meinecke P, Van Hul W : Burning down DEFECT11. Am J Med Genet 2001; 100: 331–332.
Wuyts W, Di Gennaro G, Bianco F et al: Molecular and clinical examination of an Italian DEFECT 11 family. Eur J Hum Genet 1999; 7: 579–584.
Yamamoto T, Akaboshi S, Ninomiya H, Nanba E : DEFECT 11 syndrome associated with agenesis of the corpus callosum. J Med Genet 2001; 38: E5.
Francke U, George DL, Brown MG, Riccardi VM : Gene dose effect: intraband mapping of the LDH A locus using cells from four individuals with different interstitial deletions of 11p. Cytogenet Cell Genet 1977; 19: 197–207.
Wuyts W, Van Hul W, Wauters J et al: Positional cloning of a gene involved in hereditary multiple exostoses. Hum Mol Genet 1996; 5: 1547–1557.
Gustavson HH, Annerén G, Wranne L : Two cases of 11p13 interstitial deletion and unusual clinical features. Clin Genet 1984; 26: 247–249.
Shaffer LG, Hecht JT, Ledbetter DH, Greenberg F : Familial interstitial deletion 11(p11.12p12) associated with parietal foramina, brachymicrocephaly and mental retardation. Am J Med Genet 1993; 45: 581–583.
McGaughran JM, Ward HB, Evans DGR : WAGR syndrome and multiple exostoses in a patient with del(11)(p11.2p14.2). Am J Med Genet 1995; 32: 823–824.
Potocki L, Shaffer LG : Interstitial deletion of 11(p11.2p12): a newly described contiguous gene deletion syndrome involving the gene for hereditary multiple exostoses (EXT2). Am J Med Genet 1996; 62: 319–325.
Lind T, Tufaro F, McCormick C, Lindahl U, Lidholt K : The putative tumor suppressors EXT1 and EXT2 are glycosyltranserases required for the biosynthesis of heparan sulfate. J Biol Chem 1998; 273: 26265–26268.
Stickens D, Clines G, Burbee D et al: The EXT2 multiple exostoses gene defines a family of putative tumour suppressor genes. Nat Genet 1996; 14: 25–32.
Wuyts W, Cleiren E, Homfray T, Rasore-Quartino A, Vanhoenacker F, Van HW : The ALX4 homeobox gene is mutated in patients with ossification defects of the skull (foramina parietalia permagna, OMIM 168500). J Med Genet 2000; 37: 916–920.
Mavrogiannis LA, Antonopoulou I, Baxova A et al: Haploinsufficiency of the human homeobox gene ALX4 causes skull ossification defects. Nat Genet 2001; 27: 17–18.
Bartsch O, Wagner A, Hinkel GK et al: FISH studies in 5 patients with Rubinstein–Taybi syndrome: deletions associated with polysplenia, hypoplastic left heart and death in infancy. Eur J Hum Genet 1999; 7: 748–756.
Mooser V, Maillard A, Bonny C et al: Genomic organization, fine-mapping, and expression of the human islet-brain 1 (IB1)/c-Jun-amino-terminal kinase interacting protein-1 (JIP-1) gene. Genomics 1999; 55: 202–208.
Lorenz P, Rupprecht E, Tellkamp H : An unusual type of acrocephalosyndactyly with bilateral parietooccipital encephalocele, micropenis and severe mental retardation. Am J Med Genet 1990; 36: 265–268.
Potocki L, Greenberg F, Shaffer LG : Interstitial deletion of 11(p11.12p12): a rare chromosomal syndrome with mental retardation, parietal foramina and multiple exostoses. Am J Hum Genet 1995; 57: 688.
Waeber G, Delplanque J, Bonny C et al: The gene MAPK8IP1, encoding islet-brain-1, is a candidate for type 2 diabetes. Nat Genet 2000; 24: 291–295.
Magara F, Haefliger JA, Thompson N et al: Increased vulnerability to kainic acid-induced epileptic seizures in mice underexpressing the scaffold protein Islet-Brain 1/JIP-1. Eur J Neurosci 2003; 17: 2602–2610.
Watkins H, Conner D, Thierfelder L et al: Mutations in the cardiac myosin binding protein-C gene on chromosome 11 cause familial hypertrophic cardiomyopathy. Nat Genet 1995; 11: 434–437.
Bonne G, Carrier L, Bercovici J et al: Cardiac myosin binding protein-C gene splice acceptor site mutation is associated with familial hypertrophic cardiomyopathy. Nat Genet 1995; 11: 438–440.
Honsho M, Tamura S, Shimozawa N, Suzuki Y, Kondo N, Fujiki Y : Mutation in PEX16 is causal in the peroxisome-deficient Zellweger syndrome of complementation group D. Am J Hum Genet 1998; 63: 1622–1630.
Saftig P, Hartmann D, Lullmann R et al: Mice deficient in lysosomal acid phosphatase develop lysosomal storage in the kidney and central nervous system. J Biol Chem 1997; 272: 18628–18635.
Acknowledgements
We thank the patients and families for participating in this study and Arleta Frensel and Marion Richter for excellent technical assistance. We also thank Dr Jörg Schaper for radiological evaluation and Dr Vincent Mooser for providing the IB1 BAC clone. This study was supported by Grant No. BEO 0311211, Bundesminister für Bildung und Forschung to OB and JDFRF Grant No. 1-2001-555 from the International American Diabetes Foundation and Grant No. 32-66892.01 from the Swiss National Science Foundation, to GW.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Wuyts, W., Waeber, G., Meinecke, P. et al. Proximal 11p deletion syndrome (P11pDS): additional evaluation of the clinical and molecular aspects. Eur J Hum Genet 12, 400–406 (2004). https://doi.org/10.1038/sj.ejhg.5201163
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.ejhg.5201163
Keywords
This article is cited by
-
De novo truncating variants in PHF21A cause intellectual disability and craniofacial anomalies
European Journal of Human Genetics (2019)
-
Foramina parietalia permagna: familial and radiological evaluation of two cases and review of literature
Child's Nervous System (2017)
-
Enlarged parietal foramina caused by mutations in the homeobox genes ALX4 and MSX2: from genotype to phenotype
European Journal of Human Genetics (2006)
-
Combination of WAGR and Potocki–Shaffer contiguous deletion syndromes in a patient with an 11p11.2–p14 deletion
European Journal of Human Genetics (2005)
-
Construction of a natural panel of 11p11.2 deletions and further delineation of the critical region involved in Potocki–Shaffer syndrome
European Journal of Human Genetics (2005)
