
Abstract
This article describes the identification of a novel locus (DFNB39) responsible for an autosomal recessive form of hearing loss segregating in a Pakistani consanguineous family. The hearing impaired members of this family present with profound prelingual sensorineural hearing impairment and use sign language for communications. Linkage was established to microsatellite markers located on chromosome 7q with a maximum multipoint lod score of 3.8. The region of homozygosity spans a 19 cM region that is bounded by markers D7S3046 and D7S644.
Similar content being viewed by others


Introduction
Nonsyndromic hearing loss in humans is the most genetically heterogeneous trait known. To date, over 80 loci have been localized and 30 genes have been identified for nonsyndromic hearing loss.1 Autosomal recessive nonsyndromic hearing loss (ARNSHL) is the most common form of hereditary hearing impairment and is generally characterized by a sensorineural defect.2 Identifying genes involved in hearing impairment and characterizing the proteins they encode will increase our knowledge of the molecular processes involved in the auditory system. In addition, it will improve our understanding of how these processes can become altered and lead to hearing impairment.
Material and methods
Clinical findings
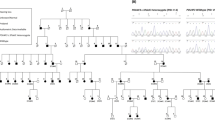
The study was initiated with the prior approval of the Institutional Review Board of Quaid-I-Azam University, Islamabad, Pakistan. Informed consent was obtained from all subjects who participated in the study. The family (pedigree 4011) described in this article resides in the Punjab province of Pakistan. Owing to strict social customs, the family members rarely marry outside of the family. Pedigree 4011 was constructed on the basis of information obtained from family members. The transmission of hearing impairment is consistent with autosomal recessive inheritance (Figure 1). The onset of hearing impairment is prelingual and most probably congenital. All affected individuals, regardless of age, display the same level of profound hearing impairment, implying that the hearing loss is not progressive. All the affected individuals use sign language for communication. There is no evidence that the hearing impairment in this kindred belongs to a syndrome or that there is gross vestibular involvement.

Drawing of pedigree 4011, which segregates DFNB39. The sexes of some of the family members have been changed to protect the anonymity of the family. Black symbols represent individuals with hearing impairment due to DFNB39. Clear symbols represent unaffected individuals. Haplotypes for the most closely linked STRPs are shown below each symbol. The markers D7S3046 and D7S644 that flank the DFNB39 region are shown in bold.
Linkage analysis
Genomic DNA was extracted from peripheral-blood samples by established procedures.3 DNA samples were obtained from 10 family members, including four individuals who had hearing impairment due to DFNB39. The samples were genotyped using microsatellite markers spaced at approximately 10 cM interval in order to localize the ARNSHL locus.
Linkage analysis was carried out under a fully penetrant autosomal recessive model with a disease allele frequency of 0.001. Two-point linkage analysis was carried out using the MLINK program of the FASTLINK computer package.4 Multipoint linkage analysis was performed using GENEHUNTER 2.1_r3 beta.5 Due to the computational limitations of GENEHUNTER and the size of the family, the pedigree was split into two halves for analysis. Lod scores were calculated for each half and then summed. For multipoint linkage analysis, marker order and map distances from the deCode genetic map6 were used.
Results
After performing linkage analysis for 50 markers, a lod score of 2.7 at θ=0 was observed for marker D7S820. In addition, all affected family members were homozygous for marker D7S820. Fine mapping of the region was carried out by genotyping 17 additional markers in this region (Table 1). Three of these markers (D7S1433, D7S611 and D7S627) were not informative for linkage and are not included in Table 1. Although none of the additional 14 markers that were genotyped exhibited a significant lod score, markers D7S524 and D7S634 also produced maximum lod scores of 2.7 at θ=0. It was observed that there was an inconsistency in the position of marker D7S820 between the Marshfield genetic map7 and both the deCode genetic map6 and the Human Genome Project – Santa Cruz sequenced-based physical map8 order (Table 1). In order to confirm that the position of marker D7S820 on the Marshfield map was potentially incorrect, Centre d'Etude du Polymorphisme Humain (CEPH) genotype data were analyzed using MAP-O-MAT.9 The support for the position of marker D7S820 on the Marshfield genetic map was low, therefore for the multipoint analysis the deCode genetic map distances and marker order were used. Those markers that were not included on the deCode genetic map were not included in the multipoint analysis (see Table 1).
Multipoint linkage analysis yielded a significant lod score of 3.8 at marker D7S2122. Three affected individuals (IV-3, IV-4, IV-5) are heterozygous for marker D7S3046 and individual (V-3) who is not hearing impaired is homozygous at D7S644 (Figure 1). Thus, the region of homozygosity is 19.2 cM according to the Marshfield genetic map,7 flanked by markers D7S3046 and D7S644. This region encompasses 17.6 Mb.8 Marker D7S3046 is not included on the deCode map, the next centromeric marker is D7S482. According to the deCode genetic map, both the region of homozygosity and the three-unit support interval is 19.8 cM and is flanked by markers D7S482 and D7S644.
Discussion
Four other loci for hearing loss DFNB4 (7q31.1), DFNB13 (7q34–36), DFNB14 (7q31) and DFNB17 (7q31) have already been mapped on the long arm of chromosome 7.1 For these three loci only the SLC26A4 gene has been identified for the DFNB4. Mutations within the SLC26A4 gene are also responsible for the Pendred syndrome10. DFNB39, which is centromeric to these loci, does not overlap with any of these loci, thus indicating the involvement of a novel gene responsible for hearing loss in kindred 4011. Further work is currently being carried out to identify the gene that is responsible for DFNB39.
References
Van Camp G, Smith RJH : Hereditary Hearing Loss Homepage (September 2002) URL: http://dnalab-www.uia.ac.be/dnalab/hhh/.
Resendes BL, Williamson RE, Morton CC : At the speed of sound: gene discovery in the auditory system. Am J Hum Genet 2001; 69: 923–935.
Grimberg J, Nawoschik S, Bellusico L McKee R, Turck A, Eisenberg A : A simple and efficient non-organic procedure for the isolation of genomic DNA from Blood. Nucleic Acid Res 1989; 17: 83–90.
Cottingham Jr RW, Indury RM, Schaffer AA : Faster sequential genetic linkage computations. Am J Hum Genet 1993; 53: 252–263.
Kruglyak L, Daly MJ, Reeve-Daly MP, Lander ES : Parametric and nonparametric linkage analysis: a unified multipoint approach. Am J Hum Genet 1996; 58: 1347–1363.
Kong A, Gudbjartsson DF, Sainz J et al: A high-resolution recombination map of the human genome. Nat Genet 2002; 31: 241–247.
Broman KW, Murray JC, Sheffield VC, White RL, Weber JL : Comprehensive human genetic maps: individual and sex specific variation in recombination. Am J Hum Genet 1998; 63: 861–869.
International Human Genome Sequence Consortium: Initial sequence and analysis of the human genome. Nature 2001; 409: 860–921.
Matise TC, Gitlin JA : MAP-O-MAT: marker-based linkage mapping on the World Wide Web. Am J Hum Genet 1999; 65: A2464.
Scott DA, Wang R, Kreman TM et al: Functional differences of the PDS gene product are associated with phenotypic variation in patients with Pendred syndrome and non-syndromic hearing loss (DFNB4). Hum Mol Genet 2000; 9: 1709–1715.
Acknowledgements
We thank the family members for their invaluable participation and cooperation. The work was funded by Quaid-I-Azam University Research Fund, the American Hearing Research Foundation and the NIH–National Institute of Deafness and other Communication Disorders Grant DC03594.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Wajid, M., Abbasi, A., Ansar, M. et al. DFNB39, a recessive form of sensorineural hearing impairment, maps to chromosome 7q11.22–q21.12. Eur J Hum Genet 11, 812–815 (2003). https://doi.org/10.1038/sj.ejhg.5201041
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.ejhg.5201041
