
Abstract
Conventional cytogenetic techniques can distinguish homologous chromosomes in a qualitative manner based upon obvious morphological features or using in situ hybridization methods that yield qualitative data. We have developed a method for quantitative genotyping of single-nucleotide variants in situ using circularizable DNA probes, so-called padlock probes, targeting two different alpha satellite repeat variants present in human chromosome 7 centromeres, and a single-nucleotide variation in alpha satellite repeats on human chromosome 15 centromeres. By using these PCR-generated padlock probes, we could quantitatively distinguish homologous chromosomes and follow the transmission of the chromosomes by in situ analysis during three consecutive generations.
Similar content being viewed by others

Introduction
All human centromeres contain alpha satellite DNA, composed of tandem repeats of approximately 171-basepair monomers organized into higher-order repeat units.1,2,3 Several chromosome-specific subfamilies of alpha satellite DNA have been identified. These subfamilies differ in the number of monomer units within each higher-order repeat and in monomer sequence.2,4 Analysis of alpha satellite DNA arrays from several homologous chromosomes has revealed polymorphisms both within and between individual centromeres, both with respect to the number of higher-order repeats and their sequence composition.5,6,7
Chromosomal heteromorphisms have been used to distinguish homologous chromosomes, and they were used to map the first gene to a human autosome–the Duffy blood group locus to chromosome 1.8 The drawback of using morphological differences is that only extreme variants, rare in the human population, can be reliably genotyped, since morphology can change depending on the chromosome preparation and banding technique used.9,10 A genetic marker should be reproducible and have many resolvable alleles to be maximally informative. The single-nucleotide polymorphisms and higher-order repeat number polymorphisms in alpha satellite DNA are potentially useful cytogenetic markers, since they are stably transmitted in the human population.11 These inter- and intrachromosomal sequence differences have been used to distinguish cytogenetically normal homologous metaphase chromosomes, and to follow the transmission of chromosomes in pedigrees.6,7,12,13 The polymorphisms have also been used to study the partitioning of homologues of chromosome 17 in normal male meiosis.14
Only a small number of techniques are available to detect single-nucleotide polymorphisms in situ. Short oligonucleotide probes have been used to analyze single-nucleotide differences in situ.6,7 This method relies upon the destabilizing effect of a single-nucleotide mismatch during stringent washing condition. Correctly matched probes remain hybridized after suitably stringent washes, while incorrectly hybridized probes dissociate. Improved selectivity is achieved with the primed in situ (PRINS) polymerization technique, where unlabeled probes are extended with labeled nucleotides in situ.15 The single-nucleotide selectivity of the method is based upon the ability of polymerases to extend matched but not 3′ mismatched primers in the extension reaction.16,17
We have previously reported a different approach to distinguish single-nucleotide differences in situ using circularizable oligonucleotide probes, so-called padlock probes.12 Padlock probes consist of two target-complementary segments at each end of an oligonucleotide probe, joined by a target-noncomplementary linker segment. Upon hybridization to the correct target sequence, the 5′ and 3′ end-segments of the probe become juxtaposed, creating a ligation substrate. The junction can be sealed with a ligase, thereby circularizing the padlock probe and rendering it physically catenated to the target sequence. The reaction is highly specific since it requires the hybridization and juxtaposition of two target-specific segments, and as the ligation reaction is strongly inhibited by mismatches at the ligation junction. The physical catenation of padlock probes to the target sequences causes the probes to remain firmly attached, even under superstringent washing conditions that remove any nonligated probes.18 Owing to their length, 70–100 nt, padlock probes are relatively expensive to synthesize chemically and require purification, particularly if detectable functions such as haptens or fluorophores are incorporated during or after synthesis. We and others have addressed this problem by developing a PCR-based method for flexible small-scale synthesis of padlock probes of greater than 100 nt, which is an approximate practical limit of chemical synthesis.13,19
Here we have designed and enzymatically synthesized padlock probes targeting two alpha satellite variants on chromosome 7,20 and a previously described single-nucleotide variation on chromosome 15.7 O'Keefe et al6 and O'Keefe and Matera7 designed short variant-specific oligonucleotide probes targeting the sequence variants present on chromosome 15, and were able to distinguish homologous chromosomes by in situ hybridization, and to follow the transmission of chromosomes from parent to offspring. However, they found this marker poorly informative. We show that this marker is more informative in a quantitative ligation-based in situ genotyping approach, since more alleles were resolved. This approach enabled us to genotype homologous chromosomes, and to follow the transmission of individual chromosomes across eight meioses in a pedigree.
Materials and methods
Oligonucleotides and cell cultures
All oligonucleotides used to synthesize padlock probes were purchased from Interactiva (Ulm, Germany) and are presented in Table 1. The CEPH family cells lines GM12547, GM12548, GM12550, GM12551, GM12552, GM12556, and GM12557 used in the in situ experiments were obtained from Coriell Cell Repositories (Camden, USA).
Synthesis of padlock probes
Padlock probes were synthesized in two consecutive amplification reactions. In the PCR, 5′-phosphorylated primer forms the 5′ target-complementary segment of the padlock probe, and the 3′ target-complementary segment of the probe is formed by the complements of the 5′ biotinylated primer. The first PCR was run with a biotinylated forward primer and a reverse primer at 0.1 μ M each (for primers see Table 1) in Taq buffer (50 mM KCl, 10 mM Tris-HCl pH 8.3, 1.5 mM MgCl2, 12.5 μg/ml BSA), with 200 μ M of each dNTP and 0.02 U/μl Taq polymerase from Applied Biosystems (Foster City, USA) using 0.1 amol λ DNA as template. The PCR was run for 35 cycles of 94°C, 55°C, and 72°C, 1 min at each temperature, before the concluding extension at 72°C for 7 min. The PCR products were gel purified and used as templates in a second PCR to create the full-length padlock probes. The second PCR was run with 0.05 μ M of a 5′-biotinylated forward primer and a phosphorylated reverse primer at 0.1 μ M using the same reaction conditions as above, except that the nucleotide concentration was 50 μ M of each dNTP and one-third of the dTTP was exchanged with either dinitrophenyl-11-dUTP (Molecular Probes, Leiden, The Netherlands) for padlock probes cen7z1 and cen15A, or digoxigenin-11-dUTP (Roche, Basel, Switzerland) for cen7z2 and cen15G. DNA was precipitated with 0.1 volume of 3 M NaAc pH 5.0 and two volumes of ice-cold 95% ethanol. The concentration of resuspended DNA was determined by spectrophotometry (GeneQuant, Amersham Biosciences, Uppsala, Sweden). DNA (4 μg) in 1 M NaCl was bound to 200 μl paramagnetic streptavidin-coated beads (Dynabeads M-280, Dynal Biotech, Oslo, Norway), prewashed with 100 μl of washing buffer (50 mM NaCl, 10 mM Tris-HCl pH 7.0), for 30 min on a test-tube rotator at room temperature. The binding solution was removed and the beads were washed three times with washing buffer before adding 50 μl of 0.15 M LiOH. After 5 min, the denaturing solution with the single-stranded padlock probes was removed to a clean test tube and neutralized with 50 μl of 0.15 M NH4Cl. The released padlock probes were precipitated with 3 μg of glycogen (Roche, Basel, Switzerland) added as carrier, resuspended in 25 μl TE-buffer, and quantitated by spectrophotometry.
In situ analysis
Metaphase chromosome preparations, spread on microscope slides, were obtained from lymphocyte cultures using standard techniques of colcemide treatment, hypotonic shock, methanol/acetic acid fixation, and RNase A treatment. DNA was denatured by placing the slides on a temperature-controlled block (GenE thermal cycler, Cambridge, UK) adding 70% formamide in 2 × SSC (1 × SSC=150 mM NaCl, 15 mM sodium citrate pH 7.0), overlaying with a cover slip, and heating the slides at 68°C for 2 min. The slides were washed in ice-cold 2 × SSC for 2 min and dehydrated in an ethanol series of 70 (ice-cold), 85, and 100% (room temperature) for 2 min each. Hybridization was performed with a probe concentration of one fmol/μl for each labeled probe in 20% formamide, 10% dextran sulfhate, 2 × SSC, and denatured sonicated salmon sperm at 0.5 μg/μl. A 24 × 50 mm2 coverslip was put over 30 μl of hybridization solution spotted onto each slide and sealed with rubber cement. The slides were incubated in a moist-chamber at 37°C overnight. The coverslip was removed and the excess of probes washed away in 37°C 2 × SSC for 5 min followed by dehydration in an ethanol series. Ligation was carried out in Tth buffer (20 mM Tris-HCl pH 7.9, 100 mM KCl, 10 mM MgCl2, 1 mM EDTA, 1 mM DTT, 0.1% Triton X-100, 1 mM NAD) supplemented with 8.7% glycerol, 0.1 μg/μl sonicated salmon sperm DNA, and 0.1 μg/μl BSA, using a Tth ligase concentration of 0.25 U/μl. The slides were placed onto a temperature-controlled block, 60 μl of ligation solution was spotted onto each slide, and a 24 × 50 mm2 coverslip was added before incubating at 55°C for 20 min. The reactions were stopped by immersing the slides in 3.3 × SSC and 50 mM EDTA at 55°C for 2 min. Unreacted padlock probes were removed by washing in 30% formamide in 2 × SSC at 42°C and 2 × SSC at 55°C for 10 min, respectively, before cooling the slides to room temperature in 2 × SSC and 0.05% Tween-20. Reacted probes were visualized by first adding mouse antidigoxigenin (Sigma-Aldrich, Munich, Germany) and rabbit antidinitrophenyl (Molecular Probes) antibodies at a concentration of 4 ng/μl in 70 μl of 2 × SSC, 0.05% Tween-20, and 1% blocking solution (Roche) for each slide and covering with a 60 × 24 mm2 coverslip. The slides were incubated at 37°C for 30 min, and then washed with gentle agitation twice in 2 × SSC and 0.05% Tween-20 for 7 min each time. The procedure above was repeated a second time with fluorophore-conjugated antibodies, goat anti-rabbit Oregon Green 488 and goat anti-mouse Alexa Fluor 594 antibodies (Molecular Probes). Finally, the slides were dehydrated in an ethanol series and then counter-stained with DAPI (Sigma-Aldrich) 100 ng/μl in Vectashield mounting medium (Vector Laboratories Inc., Burlingame, USA) under a 50 × 24 mm2 coverslip. The slides were viewed under a microscope (Axioskop, Zeiss, Oberkochen, Germany) and at least 20 metaphase images/genotyped individual were captured with a CCD camera (CE 200A, Photometrics GmBH, Munich, Germany). The images were processed using the Vysis software Quips FISH (Downers Grove, USA). They were normalized before the quantitative analysis by subtracting background noise and setting the strongest signal to 100%. Normalized fluorescence signal values were obtained by dividing the mean signal intensities from 20 metaphase images of the test sample with the corresponding mean signal intensities from the strongest allele in 20 metaphase images obtained from the internal standard sample. Cells from individual 12 547 were used as the internal standard, and were spread onto the same experimental slide and subjected to the same experimental procedure and analysed as the test cells.
Results
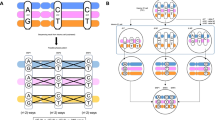
For each chromosome locus, a pair of 146 nt long padlock probes, specific for the two alpha satellite DNA variants of chromosomes 7 and 15, were synthesized by PCR incorporating either dinitrophenyl-11-dUTP or digoxigening-11-dUTP (see Table 1 for sequences). We estimate that 10–20 modified nucleotides were incorporated per probe molecule. The two probe pairs were added simultaneously, and hybridized and ligated on metaphase preparations. The probes were visualized using two layers of fluorescence-labeled antibodies. The staining patterns for chromosome 15 from three members of the family are shown in Figure 1a. By quantifying the red and green signals present on the centromeres of chromosome 15, the homologous centromeres could be distinguished (Figure 1b). Both pairs of the chromosomes 7 and 15 cytogenetic markers were combined informative for all seven tested individuals. This enabled us to genotype and follow the transmission of centromeres on chromosomes 7 and 15 through three generations (Figure 2).

PCR-generated padlock probes distinguish individual chromosomes 15 through quantitative fluorescence analysis. (a) Padlock probes clearly distinguish the different chromosome 15 genotypes present in three related individuals in CEPH pedigree 66. In situ hybridization was performed with cen15G and cen15A padlock probes labeled with digoxigenin and dinitrophenyl, respectively. Probes were detected with fluorophore-conjugated antibodies. (b) The relative red and green intensities of the chromosome 15 signals in (a) were quantified from 20 metaphase images. The mean signal intensities are shown for the three individuals, and the allele identities are according to Figure 2. The error bars denote the standard error of the mean.

Mendelian inheritance of the chromosomes 7 and 15 centromeres through three generations. The numbers below each individual correspond to the cell culture number from the Coriell cell repository. The different alleles of chromosomes 7 and 15 are denoted with numbers.
To estimate how many alleles can be distinguished using this quantitative approach, we plotted all the chromosome 7 alleles in the family and joined shared alleles according to their segregation in the family (Figure 3). Although the cen7z1 marker added little resolving power, four alleles could be discerned mostly based on the difference in cen7z2 signal intensity. To further enhance the reproducibility and the quantitative resolution of the method, we normalized the signals to an internal quantification standard. The normalization was performed by dividing the mean fluorescence values from the test sample with the corresponding mean fluorescence values obtained from cells of a reference sample, present on a different location on the same slide. To evaluate the precision of the procedure we compared the in situ genotyping results from two individuals differing at the chromosome 7 loci while sharing centromeric alleles at the chromosome 15 loci (Figure 4). The normalized signals from the shared alleles clearly overlapped even if they were present in different cell lines, while the alleles that differed exhibited significantly different signal intensities (Figure 4b).

The number of resolvable alleles of the genetic in situ markers cen7z1 and cen7z2. All chromosome 7 centromeric alleles were plotted according to their respective relative fluorescence intensities. The segregation information from Figure 2 was used to join shared alleles (broken line). The allele groups are labeled according to their identity in Figure 2. Singletons (alleles 1 and 4) were not included in the allele groups.

The reproducibility of quantitative fluorescence analysis of padlock probes is improved by normalizing the signals to a reference sample present on the same slide. (a) Mean red and green signal intensities and (b) normalized signals from 20 metaphase images from individuals 12 550 (filled symbols) and 12 551 (open symbols) were plotted. Signals from chromosomes 7 and 15 are denoted by circles and squares, respectively. The allele identity numbers are according to Figure 2. Relative standard error of the mean is shown by the error bars.
Discussion
As shown here, padlock probes provide quantitative data, resulting in precise genotyping of centromeres of chromosomes 7 and 15. Padlock probes give rise to signals that can be reliably quantified in contrast to the reported results obtained using short variant-specific oligonucleotide hybridization probes.6,7 This difference may be because of the sequence discriminating power of the ligation reaction, and the physical catenation of the circularized probe to the target sequence, which renders reacted probes resistant to superstringent washes, thereby minimizing loss of signal. Short variant-specific oligonucleotide probes require stringent washes in order to be selective, inevitably resulting in some loss of probes with a concomitant decrease of signal. The cohybridization and ligation of padlock probes specific for alternate sequence variants serve to decrease the variability inherent to seq uentially performed reactions such as dual color PRINS.21
The results we obtained from the relative fluorescence determinations allowed us to discern at least four alleles in a set of seven related individuals, based on the fluorescence from one marker (Figure 3). If a centromer has two informative markers, like the centromeric in situ markers for chromosomes 13 and 21,12,13 then it should be possible to distinguish at least 16 different alleles on the basis of relative fluorescence signals. Qualitative markers are limited to positive or negative scores and can consequently only identify two different alleles using one marker, and four alleles using two markers. To further increase the resolution of different alleles, an internal standard can be used to normalize the fluorescence signals. One way to normalize fluorescence signals is to relate the fluorescence measurements to signals from externally added fluorescent beads with well-characterized fluorescence intensities.22 We chose to normalize the fluorescent signals by reference to a second sample, in situ genotyped on the same slide as the test sample. This normalization method has previously been used to quantify human telomere size.23 The normalized results clearly demonstrate that the method is reproducible and that interexperimental variations can be minimized using an internal fluorescence standard. The normalization procedure may therefore increase the number of alleles that can be discerned compared to when using relative fluorescence data.
Padlock probes are well suited for multiplexing because of their stringency and low crossreactivity.18,24 However, the number of colors that can be used in an in situ experiment is limited by the number of haptens available, and that can be incorporated in the probes. We have, however, successfully been able to directly label the probes by incorporating fluorescence-labeled nucleotides during PCR, which increases the multiplexing possibilities substantially (unpublished results). Multiplexing is also limited by the number of colors that can be visualized with standard fluorescence microscope systems. Most systems can only separate seven colors at the most. This can be overcome by introducing ratio- or combinatorial labeling of the probes.25,26
A number of single-nucleotide variations of alpha-satellite sequences are known today, currently limited to chromosomes 13, 15, 17 and 21.6,7,12 Centromeres containing two different alpha-satellite repeats can also be distinguished using padlock probes, as shown here with chromosome 7. Taking to account the large amount of sequence data available in databases, and the sequence variability of alpha-satellite repeats, it is likely that there exist suitable combinations of alpha-satellite repeats or single-nucleotide polymorphisms to detect all chromosomes.
In conclusion, padlock probes are useful tools to investigate sequence variation in centromeres in situ. They can be used to establish parent of origin in dicentric chromosomes or chromosomes with a balanced translocation, and to investigate if nondisjunction has occurred in meiosis I or II in studies of aneuploidy.
References
Manuelidis L, Wu JC : Homology between human and simian repeated DNA. Nature 1978; 276: 92–94.
Willard HF : Chromosome-specific organization of human alpha satellite DNA. Am J Hum Genet 1985; 37: 524–332.
Waye JS, Willard HF : Nucleotide sequence heterogeneity of alpha satellite repetitive DNA: a survey of alphoid sequences from different human chromosomes. Nucleic Acids Res 1987; 15: 7549–7569.
Jorgensen AL, Bostock CJ, Bak AL : Chromosome-specific subfamilies within human alphoid repetitive DNA. J Mol Biol 1986; 187: 185–196.
Durfy SJ, Willard HF : Patterns of intra- and interarray sequence variation in alpha satellite from the human X chromosome: evidence for short-range homogenization of tandemly repeated DNA sequences. Genomics 1989; 5: 810–821.
O'Keefe CL, Warburton PE, Matera AG : Oligonucleotide probes for alpha satellite DNA variants can distinguish homologous chromosomes by FISH. Hum Mol Genet 1996; 5: 1793–1799.
O'Keefe CL, Matera AG : Alpha satellite DNA variant-specific oligoprobes differing by a single base can distinguish chromosome 15 homologs. Genome Res 2000; 10: 1342–1350.
Donahue RP, Bias WB, Renwick JH, McKusick VA : Probable assignment of the Duffy blood group locus to chromosome 1 in man. Proc Natl Acad Sci USA 1968; 61: 949–955.
Sherman SL, Takaesu N, Freeman SB et al: Trisomy 21: association between reduced recombination and nondisjunction. Am J Hum Genet 1991; 49: 608–620.
Lorber BJ, Grantham M, Peters J, Willard HF, Hassold TJ : Nondisjunction of chromosome 21: comparisons of cytogenetic and molecular studies of the meiotic stage and parent of origin. Am J Hum Genet 1992; 51: 1265–1276.
Wevrick R, Willard HF : Long-range organization of tandem arrays of alpha satellite DNA at the centromeres of human chromosomes: high-frequency array-length polymorphism and meiotic stability. Proc Natl Acad Sci USA 1989; 86: 9394–9398.
Nilsson M, Krejci K, Koch J, Kwiatkowski M, Gustavsson P, Landegren U : Padlock probes reveal single-nucleotide differences, parent of origin and in situ distribution of centromeric sequences in human chromosomes 13 and 21. Nat Genet 1997; 16: 252–225.
Antson DO, Isaksson A, Landegren U, Nilsson M : PCR-generated padlock probes detect single nucleotide variation in genomic DNA. Nucleic Acids Res 2000; 28: E58.
O'Keefe CL, Griffin DK, Bean CJ, Matera AG, Hassold TJ : Alphoid variant-specific FISH probes can distinguish autosomal meiosis I from meiosis II non-disjunction in human sperm. Hum Genet 1997; 101: 61–66.
Koch JE, Kolvraa S, Petersen KB, Gregersen N, Bolund L : Oligonucleotide-priming methods for the chromosome-specific labelling of alpha satellite DNA in situ. Chromosoma 1989; 98: 259–265.
Pellestor F, Girardet A, Andreo B, Charlieu JP : A polymorphic alpha satellite sequence specific for human chromosome 13 detected by oligonucleotide primed in situ labelling (PRINS). Hum Genet 1994; 94: 346–348.
Pellestor F, Girardet A, Lefort G, Andreo B, Charlieu JP : Use of the primed in situ labelling (PRINS) technique for a rapid detection of chromosomes 13, 16, 18, 21, X and Y. Hum Genet 1995; 95: 12–17.
Nilsson M, Malmgren H, Samiotaki M, Kwiatkowski M, Chowdhary BP, Landegren U : Padlock probes: circularizing oligonucleotides for localized DNA detection. Science 1994; 265: 2085–2088.
Myer SE, Day DJ : Synthesis and application of circularizable ligation probes. Biotechniques 2001; 30: 584–58, 590, 592–593.
Waye JS, England SB, Willard HF : Genomic organization of alpha satellite DNA on human chromosome 7: evidence for two distinct alphoid domains on a single chromosome. Mol Cell Biol 1987; 7: 349–356.
Coignet L, Girardet A, Andreo B, Charlieu JP, Pellestor F : Double and triple in situ chromosomal labeling of human spermatozoa by PRINS. Cytogenet Cell Genet 1996; 73: 300–330.
Zijlmans JM, Martens UM, Poon SS et al: Telomeres in the mouse have large inter-chromosomal variations in the number of T2AG3 repeats. Proc Natl Acad Sci USA 1997; 94: 7423–7428.
Krejci K, Koch J : Improved detection and comparative sizing of human chromosomal telomeres in situ. Chromosoma 1998; 107: 198–203.
Nilsson M, Baner J, Mendel-Hartvig M et al: Making ends meet in genetic analysis using padlock probes. Hum Mutat 2002; 19: 410–415.
Speicher MR, Gwyn Ballard S, Ward DC : Karyotyping human chromosomes by combinatorial multi-fluor FISH. Nat Genet 1996; 12: 368–375.
Tanke HJ, Wiegant J, van Gijlswijk RP et al: New strategy for multi-colour fluorescence in situ hybridisation: COBRA: COmbined Binary RAtio labelling. Eur J Hum Genet 1999; 7: 2–11.
Acknowledgements
We thank Dr Francis Barany for kindly providing the Tth DNA ligase used in this study. This work was supported by grants from the Beijer, the Wenner-Gren, and the Borgstrom Foundations, the Foundation for Medical Research in Uppsala, Svenska Lakar Sallskapet, the Swedish Research Council, and the Wallenberg Consortium North for Functional Genomics.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Antson, DO., Mendel-Hartvig, M., Landegren, U. et al. PCR-generated padlock probes distinguish homologous chromosomes through quantitative fluorescence analysis. Eur J Hum Genet 11, 357–363 (2003). https://doi.org/10.1038/sj.ejhg.5200966
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.ejhg.5200966
Keywords
This article is cited by
-
Lock and roll: single-molecule genotyping in situ using padlock probes and rolling-circle amplification
Histochemistry and Cell Biology (2006)
-
In situ genotyping individual DNA molecules by target-primed rolling-circle amplification of padlock probes
Nature Methods (2004)
