
Abstract
Anophthalmia or microphthalmia occur in approximately one in 10 children who have severe visual impairment. These eye malformations are often of unknown aetiology, but can be inherited in autosomal dominant, recessive or X-linked forms, and can also occur in association with specific chromosome abnormalities. Four children are described in the medical literature with microphthalmia or anophthalmia in association with chromosome rearrangements involving distal 3q, suggesting the presence of a micro/anophthalmia gene in this region. We have identified two further patients with micro/anophthalmia and chromosome rearrangements involving 3q26→3q27 and identified a 6.7 MB common deleted region. Patient 1 had multiple abnormalities including bilateral anophthalmia, abnormalities of the first and second cranial nerves and partial absence of the corpus callosum. His karyotype was 46,XY,del(3)(q26.33q28). Patient 2 had right anophthalmia and left extreme microphthalmia. Her karyotype was 46,XX,del(3)(q26.33q28)t(3;7)(q28;q21.1). Both patients had intrauterine growth retardation (IUGR) and strikingly similar dysmorphic facies consisting of bossed forehead, downward-slanting palpebral fissures, grooved bridge of the nose, prominent low-set ears, small down-turned mouth and small mandible. We identified BAC clones mapping to distal 3q from the ENSEMBL and NCBI Entrez databases. These BAC clones were used as fluorescence in situ hybridisation (FISH) probes to identify the minimum deleted region common to both patients. This interval, between clones RPC11-134F2 and RPC11-132N15, was estimated to be 6.7 MB. We conclude that there is an anophthalmia locus within this interval. Candidate genes mapping to this region include Chordin and DVL3, a homologue of the Drosophila Dishevelled gene.
Similar content being viewed by others


Introduction
Microphthalmia affects approximately 1.8 individuals per 10 000,1 while anophthalmia, at the extreme end of the microphthalmia spectrum, occurs with a frequency of 0.3–0.6 individuals per 10 000.1,2 The defect is variable in severity and is present bilaterally in only 50% of cases. There is significant recurrence risk within families, whether the micro/anophthalmia is unilateral or bilateral.3 The most severe form of disease, bilateral anophthalmia or bilateral extreme microphthalmia, is rare, and has been estimated to occur with a maximum incidence of 1.4 per 100 000 individuals (D FitzPatrick, unpublished data). Both anophthalmia and microphthalmia can occur in syndromic form, in conjunction with other congenital abnormalities, or in isolation. It has been estimated by an international survey that microphthalmia or anophthalmia are found in 13% of all children with severe visual impairment.4
Although many cases of microphthalmia or anophthalmia are sporadic, there is proven genetic aetiology in a proportion of cases. Syndromic forms of microphthalmia are reported with established Mendelian patterns of inheritance: these include X-linked Lenz microphthalmia5 and autosomal recessive Waardenberg type anophthalmia.6 There are also case reports of families showing autosomal dominant, autosomal recessive and X-linked forms of non-syndromic anophthalmia or microphthalmia.7,8,9 Linkage analysis in these families has led to the mapping of loci at 15q12-q15,7 14q328 and Xq279 and the identification of a gene for recessive microphthalmia, CHX10, at 14q24.10 Microphthalmia or anophthalmia are also seen in association with chromosome aneuploidy, particularly trisomy 13, and specific chromosome rearrangements, particularly deletions involving 14q22. SIX6 has been identified as a potential candidate on 14q.11
Several cases in the medical literature point to an association between anophthalmia and microphthalmia and rearrangements of distal 3q. Of five patients reported with a terminal deletion of 3q,12,13,14,15,16 one had microphthalmia12 and one had anophthalmia.13 All five cases had additional dysmorphic features. However, there have also been two reports of isolated cases of micro/anophthalmia associated with apparently balanced translocations involving 3q27. A patient with bilateral anophthalmia associated with t(3;11)(q27;p11.2) was reported by Driggers et al.17 and one with bilateral microphthalmia and colobomatous cysts, t(3;5)(q27;q11.2) by Kurbasic et al.18
We report two new patients with bilateral micro/anophthalmia as well as other dysmorphic features in association with chromosomal deletions involving 3q26-28. Using fluorescence in situ hybridisation we have delineated a common region of the deletion in our patients, giving a candidate interval of 6.7 MB for the putative anophthalmia locus on 3q26→28.
Methods
Patients
Both patients presented to their regional genetics centre with an unknown dysmorphic syndrome associated with bilateral micro/anophthalmia. Both underwent routine chromosome analysis, using standard cytogenetic techniques, as part of routine clinical assessment.
Acquisition of clones
BAC clones mapping to the appropriate region of 3q were identified from the NCBI Genome database (http://www.ncbi.nlm.nih.gov/genome/guide/human) and the Ensembl database (http://www/ensembl.org/Homo_Sapiens/). Clones were chosen at approximately 0.5 MB intervals. These were purchased from the BacPac Resource Centre (http://www.chori.org/bacpac/).
Fluorescence in situ hybridisation (FISH)
Single BAC colonies were inoculated into 3 ml LB/chloramphenicol and cultured overnight, followed by a standard Quiagen™ miniprep procedure. Five hundred ng of clone DNA was labelled with either biotin or digoxigenin by nick translation (BioNick Kit or Nick Translation System respectively, both BRL Life Technologies).
Per FISH slide, 50 ng of each probe plus 50–200× (w/w) Cot-1 DNA was dried down and re-suspended in hybridisation buffer (50% formamide, 2×SSC, 1% Tween-20, 20% dextran sulphate, pH 7). Chromosome preparations from lymphocyte or fibroblast cultures were prepared by standard cytogenetic techniques. Probe and target DNAs were co-denatured by heating to 72°C for 6 min and then hybridised at 37°C overnight (approximately 16 h). Signal detection was by alternate layers of Texas red-conjugated avidin and biotinylated anti-avidin for biotin labelled probes, and a single layer of fluorescein isothiocyanate-conjugated anti-digoxigenin (FITC anti-digoxigenin) for digoxigenin-labelled probes. Slides were counter-stained with 4,6-diamidino-2-phenylindole (DAPI) in anti-fade solution. Analysis was carried out using an epifluorescence microscope with CCD camera and image analysis system (Applied Imaging, UK). Clone position was verified using high resolution chromosome preparations.
Estimation of size of candidate interval
A virtual contig of BAC clones was constructed using the Human FPC Database at the Genome Sequencing Centre, Washington University (St Louis, MO, USA) (http://www.genome.wustl.edu/gsc/human/details.shtml).19 Existing contigs available on this site were supplemented using HindIII fingerprint data to identify overlapping clones.
Identification of candidate genes
Genes mapping to our candidate interval were identified from the ENSEMBL and NCBI genome databases.
Results
Patients
The clinical features of our two patients are compared with those of other reported cases in Table 1. Patient 1 was male, the second child of non-consanguineous parents. He was born at 37 weeks by caesarian section. Birth weight (1960 g), length (43 cm) and head circumference (30 cm) were all below the 3rd centile. Multiple abnormalities were noted at birth. He had bilateral anophthalmia, although eyelids and eyelashes were present, a dysmorphic face, midline cleft palate, posteriorly placed tongue and micrognathia. The penis was very small with a hypoplastic scrotum and bilateral cryptorchidism. Cranial ultrasound and MRI showed bilateral anophthalmia and agenesis of the corpus callosum. The patient died at 40 days of age from respiratory failure. Post mortem examination confirmed the clinical findings and also demonstrated a small posterior cleft of the larynx extending to the vocal folds, 13 pairs of ribs and a cleft odontoid peg.
Examination of the central nervous system confirmed anophthalmia with absent optic nerves and chiasm, absence of the first cranial nerve on the right and partial absence of the corpus callosum. The brain was small with thickened foci of cortex showing random orientation of neurons. There was hyperplasia of the pontine nuclei.
Routine analysis showed an apparently terminal deletion of 3q. However, follow-up FISH studies with a probe for the subtelomeric region of 3q, and other probes known to map to 3q28, showed that the deletion was in fact interstitial. The karyotype was 46,XY,del(3)(q26.33q28).
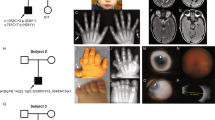
Patient 2 was female, the first child of unrelated parents, born at term with birth weight (1730 g) and head circumference (31.8 cm) also under the 3rd centile. She was initially thought to have bilateral anophthalmia but subsequent ophthalmic examination showed right anophthalmia and severe microphthalmia on the left. She had a very similar face to Patient 1. She is shown in Figure 1. The only other abnormality detected was mild aortic stenosis that did not require treatment.

Face of Patient 2.
Routine chromosome analysis showed an unusual karyotype consisting of a de novo translocation with an associated interstitial deletion at the 3q breakpoint: 46,XX,del(3) (q26.33q28)t(3;7)(q28;q21.1). This is shown in Figure 2.

The results of FISH using BAC clones mapping to distal 3q in Patient 2: (a) Ideogram showing karyotype. Red and red/black arrows show proximal and distal breakpoints of deleted region of chromosome 3. Red/black arrow also shows breakpoint of translocated section from chromosome 3. Black arrow indicates translocation breakpoint on chromosome 7. (b) FISH study on Patient 2. Closed arrow indicates normal chromosome 3 showing green signal from 430L16 and red signal from 757O23 (Yellow signal arises from the overlap of red and green signals). Open arrow shows der(7) onto which the green 430L16 signal has been translocated. Arrowhead indicates der(3) which has no signal. There is no second red signal, indicating that one copy of 757O23 has been deleted.
FISH mapping
The results of FISH using BAC clones mapping to distal 3q are summarised in Figure 3. In both patients RP11-134F2 (at 3q26.33) was the most proximal probe present. Patient 1 had a larger deletion with the distal breakpoint in 3q28 flanked by RP11-10K11 which was present distally. Patient 2 had a distal breakpoint at 3q27.3 with RP11-132N15 the first probe present on the translocated segment. The common deletion region delineated to date is therefore equivalent to the entire deletion in Patient 2, which falls between RP11-134F2 and RP11-132N15.

Representation of the candidate interval on chromosome 3q. Nondeleted probes shown by a closed black circle, deleted probes by a dash. The scale at the bottom indicates the position of each probe along the Human FPC Database contig 3019. 132N15 is labelled in parentheses because it does not appear on contig 3019. The position of Chrodin and DVL3 is shown.
Estimation of candidate region
The Human FPC Database at the Genome Sequencing Center, Washington University, St Louis, MO, USA, places all our probes with the exception of RP11-132N15 on virtual contig 3019. The order of the probes on contig 3019 corresponds with the order established by FISH (Figure 3). The distance between RP11-134F2 and RP11-132N15 was estimated to be 6.7 MB.
Candidate genes located in our region
Over 50 genes mapping to our candidate region are identified from the Human Genome Sequencing Project. These include Dishevelled 3 (DVL3) a homologue of the Drosophila dishevelled (dsh) gene20 and Chordin (CHRD) a developmental protein known to be involved in dorsoventral patterning in the mouse.21
Discussion
We have identified two new patients with deletions of 3q26-q28 and bilateral micro/anophthalmia, in association with other dysmorphic features. We have shown that the deletions overlap and that the minimum common deleted interval is approximately 6.7 MB in size.
In addition to eye defects, both patients had marked IUGR, microcephaly and very similar dysmorphic facies. Patient 1 had further congenital abnormalities similar to those described in the medical literature in other patients with microphthalmia/anophthalmia and distal 3q rearrangements. A comparison of features between our cases and those previously reported is given in Table 1.
We propose that there is a recognisable phenotype associated with deletions of distal 3q consisting of micro/anophthalmia, IUGR, the distinctive facies described above, central nervous system anomalies especially of midbrain structures, cleft palate and 13 pairs of ribs. Both of our patients had a more complex phenotype than the patient described with an apparently balanced (3;11) translocation who had micro/anophthalmia with few additional features.17 This suggests that a proportion of the additional features in our patients were caused by deletion of contiguous genes. A number of patients with micro/anophthalmia in association with other associated features such as growth restriction and microcephaly have apparently normal chromosomes. It is possible that a proportion of such patients have a mutation in a single gene or a submicroscopic deletion involving the same region of 3q.
Six cases of microphthalmia or anophthalmia in association with deletions/rearrangements of chromosome 3q have now been reported (Table 1). In three of these cases, the two we have analysed here and the patient reported by Driggers et al.17 with a (3;11) translocation, FISH has confirmed a common deleted chromosome region on 3q (D FitzPatrick, unpublished data). As micro/anophthalmia is an unusual and distinctive malformation, these data are strongly suggestive of a micro/anophthalmia locus within this common deleted region of 3q. Haploinsufficiency for the gene at this locus might be sufficient to cause the micro/anophthalmia phenotype. If this is the case, the three patients described in the literature with a terminal deletion of 3q but no eye abnormalities14,15,16 are presumed to have breakpoints which do not disrupt this gene. Our patient 1 was thought to have a terminal deletion of 3q on initial chromosome analysis, but proved to have an interstitial deletion. If the cases reported by Jokiaho et al.,16 Sargent et al.14 and Brueton et al.15 were true terminal deletions of 3q, the deleted chromosomal regions may not overlap at all. Alternatively, the affected patients may already have a mutation in the gene on the non-deleted chromosome, which is ‘unmasked’ by the loss of the second copy by chromosomal deletion.
If the locus on 3q is responsible for cases of inherited or ‘sporadic’ micro/anophthalmia in the absence of a chromosomal rearrangement it is not possible to say, from our work, whether this would be inherited as an autosomal recessive or an autosomal dominant trait.
A large number of genes and ESTs map to our 6.7 MB candidate region. Two of these genes, DVL3 and Chordin, are candidates for anophthalmia by virtue of their proposed function or pattern of expression. DVL3 is a homologue of the Drosophila segment polarity gene dishevelled (dsh) which is putatively involved with neural and heart development via wnt (vertebrate Wingless homologue) signal transduction.22 Early eye development involves patterning along polarity axes,23,24 and there is evidence that wnt signalling is involved via the frizzled receptor family in Drosophila23 and Xenopus.25 Mouse DLV3 is expressed in every embryonic tissue between 7.5 and 9.5 days of development but is maximally expressed by 10.5 days throughout the developing central nervous system as well as in branchial arches and heart.20 CHRD encodes chordin, a bone morphogenetic protein (BMP) antagonist that is involved in neural induction in Xenopus, possibly in specifying anterior neural structures including eyes.24 In mice it is also thought to play a role in patterning of the early embryo, primarily in establishing the dorsoventral axis.21 Targeted inactivation of chordin in mice leads to defects in ear, pharynx and cardiovascular organisation.26 Mice that were double homozygous mutants for chordin and noggin had absent forebrain, eyes, nasal placodes and other facial structures, including agnathia. Hindbrain was relatively unaffected.26
In conclusion, our patients have confirmed the association between rearrangements of distal 3q and micro/anophthalmia. Using FISH, we have delineated a common deleted chromosomal region of 6.7 MB at 3q26.33–3q27.3. These data suggest that a locus for micro/anophthalmia lies within this interval.
References
Stoll C, Alembik Y, Dott B, Roth MP . Congenital eye malformations in 212,479 consecutive births Ann Genet 1997 40: 122–128
Clementi M, Turolla L, Mammi I, Tenconi R . Clinical anophthalmia: an epidemiological study in northeast Italy based on 368,256 consecutive births Teratology 1992 46: 551–553
Morrison D, Fitzpatrick D, Hanson I et al. National study of microphthalmia, anophthalmia, and coloboma (MAC) in Scotland: investigation of genetic aetiology J Med Genet 2002 39: 16–22
Hornby SJ, Gilbert CE, Rahi JK et al. Regional variation in blindness in children due to microphthalmia, anophthalmia and coloboma Ophthalmic Epidemiol 2000 7: 127–138
Traboulsi EI, Lenz W, Gonzales-Ramos M, Siegel J, Macrae WG, Maumenee IH . The Lenz microphthalmia syndrome Am J Ophthalmol 1998 105: 40–45
Cogulu O, Ozkinay F, Gunduz C, Sapmaz G, Ozkinay C . Waardenburg anophthalmia syndrome: report and review Am J Med Genet 2000 90: 173–174
Morle L, Bozon M, Zech JC et al. A locus for autosomal dominant colobomatous microphthalmia maps to chromosome 15q12-15 Am J Hum Genet 2000 67: 1592–1597
Bessant DA, Anwar K, Khaliq S et al. Phenotype of autosomal recessive congenital microphthalmia mapping to chromosome 14q32 Br J Ophthalmol 1999 83: 919–922
Graham CA, Redmond RM, Nevin NC . X-linked clinical anophthalmos. Localization of the gene to Xq27-Xq28 Ophthalmic Paediatric Genet 1991 12: 43–48
Percin EF, Ploder LA, Yu JJ et al. Human microphthalmia associated with mutations in the retinal homeobox gene CHX10 Nat Genet 2000 25: 397–401
Gallardo ME, Lopez-Rios J, Fernaud-Espinosa I et al. Genomic cloning and characterization of the human homeobox gene SIX6 reveals a cluster of SIX genes in chromosome 14 and associates SIX6 hemizygosity with bilateral anophthalmia and pituitary anomalies Genomic 1999 61: 82–91
Alvarez-Arratia A, Rivera H, Moller M, Valdividia A, Vigueras A, Cantu J . De Novo del(3)(q2800) Ann Genet 1984 27: 109–111
Chitayat D, Babul R, Silver MM et al. Terminal deletion of the long arm of chromosome 3 [46,XX,del(3)(q27→qter)] Am J Med Genet 1996 61: 45–48
Sargent C, Burn J, Baraitser M, Pembrey ME . Trigonocephaly and Opitz C syndrome J Med Genet 1985 22: 39–45
Brueton LA, Barber JC, Huson SM, Winter RM . Partial monosomy 3q in a boy with short stature, developmental delay, and mild dysmorphic features J Med Genet 1989 26: 729–730
Jokiaho I, Salo A, Niemi KM, Blomstedt GC, Pihkala J . Deletion 3q27→3qter in an infant with mild dysmorphism, parietal meningocele, and neonatal milaria rubra-like lesions Hum Genet 1989 83: 302–304
Driggers RW, Macri CJ, Greenwald J et al. Isolated bilateral anophthalmia in a girl with an apparently balanced de novo translocation: 46,XX,t(3;11)(q27;p11.2) Am J Med Genet 1999 87: 201–202
Kurbasic M, Jones FV, Cook LN . Bilateral microphthalmia with colobomatous orbital cyst and de-novo balanced translocation t(3;5) Ophthalmic Genetics 2000 21: 239–242
Le Hellard S, Semple CAM, Morris SW, Porteous DJ, Evans KL . Physical mapping integrating computational and molecular genetic data Ann Hum Genet 2001 65: 221–228
Bui TD, Beier DR, Jonssen M et al. cDNA cloning of a human dishevelled DVL-3 gene, mapping to 3q27 and expression in human breast and colon carcinomas Biochem Biophys Res Commun 1997 239: 510–516
Pappano WN, Scott IC, Clark TG, Eddy RL, Shows TB, Greenspan DS . Coding sequence and expression patterns of mouse chordin and mapping of the cognate mouse chrd and human CHRD genes Genomics 1998 52: 236–239
Tsang M, Lijam N, Yang Y, Beier DR, Wynshaw-Boris A, Sussman DJ . Isolation and characterization of mouse dishevelled-3 Devel Dynamics 1996 207: 253–262
Mlodzic M . Plantar polarity in the Drosophila eye: a multifaceted view of signalling specificity and cross talk EMBO J 1999 18: 6873–6879
Chow RL, Lang RA . Early eye development in vertebrates Ann Rev Cell Dev Biol 2001 17: 255–296
Rasmussen JT, Deardoff MA, Change T et al. Regulation of eye development by frizzled signalling in Xenopus PNAS 98: 3861–3866
Bachiller D, Klingensmith J, Kemp C et al. The Organiser factors Chordin and Noggin are required for mouse forebrain development Nature 2000 403: 658–661
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Male, A., Davies, A., Bergbaum, A. et al. Delineation of an estimated 6.7 MB candidate interval for an anophthalmia gene at 3q26.33-q28 and description of the syndrome associated with visible chromosome deletions of this region. Eur J Hum Genet 10, 807–812 (2002). https://doi.org/10.1038/sj.ejhg.5200890
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.ejhg.5200890
Keywords
This article is cited by
-
Association of a de novo16q copy number variant with a phenotype that overlaps with Lenz microphthalmia and Townes-Brocks syndromes
BMC Medical Genetics (2009)
-
Separation of the PROX1 gene from upstream conserved elements in a complex inversion/translocation patient with hypoplastic left heart
European Journal of Human Genetics (2009)
-
Mutations in SOX2 cause anophthalmia
Nature Genetics (2003)
