
Abstract
Spinal neurofibromas are found in up to 38% of NF1 patients. However, they cause clinical implications only in about 5% of the patients. In contrast, multiple symptomatic spinal neurofibromas are the main clinical finding in patients with familial spinal neurofibromatosis. Familial spinal neurofibromatosis has been considered to be a distinct clinical form of neurofibromatosis. Linkage analysis in two families and identification of a NF1 gene mutation in a third family strongly associate spinal neurofibromatosis with the NF1 gene. We describe a NF1 patient who satisfies the NIH diagnostic criteria and has severe spinal involvement with bilateral spinal root neurofibromas at every level. A recurrent splice site mutation (IVS19b-3C>G) was identified in the NF1 gene in the patient. We discuss the possibility that the clinical picture of this patient represents an additional example of spinal neurofibromatosis. By comparison of the clinical expression of NF1 in this patient and that in another patient with the identical mutation the hypothesis that spinal neurofibromatosis is associated with a particular mutation is highly unlikely. The involvement of other genes linked to the NF1 gene or modifying genes is currently the most likely explanation for the clinical phenotype of spinal neurofibromatosis.
Similar content being viewed by others

Introduction
Neurofibromatosis type 1 (NF1 (MIM 162200)) is one of the most common autosomal dominant disorders, affecting approximately 1 in 3500 individuals. A hallmark of NF1 is the extreme heterogeneity in clinical expression, even among related individuals carrying the same mutation.1,2 Each of the classical signs of NF1 such as café au lait spots (CLS), iris Lisch nodules and cutaneous neurofibromas are found in more than 90% of the patients. Other features are found less frequently.
Spinal neurofibromas with clinical implications are reported in only about 5% of NF1 patients.3,4,5 This estimate may be too low, since, MRI studies in randomly selected asymptomatic NF1 patients show that spinal neurofibromas are found in up to 38% of patients.4,6,7,8 Tonsgard et al4 report that paraspinal masses particularly of the thoracic region frequently involve symmetrically multiple vertebral levels. However, multiple neurofibromas symmetrically affecting all vertebral levels of the cranial, thoracic and lumbar spine have so far only been described in patients with familial spinal neurofibromatosis (FSNF (MIM 162210)). FSNF with multiple spinal cord neurofibromas as its main clinical feature has, therefore, been considered an alternate form of neurofibromatosis.9,10,11
In this report we present a patient severely affected by spinal neurofibromas carrying a recurrent splice site mutation and fulfilling the NIH criteria for NF1.
Clinical report
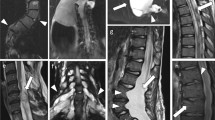
At the age of 25 years a female patient (NF1399) presented with incomplete paraparesis in arms and legs, and a sensory deficit below the craniocervical region. Magnetic resonance tomography (MRT) revealed severe spinal involvement as multiple paraspinal nerve sheath tumours were found symmetrically on either side of each segment of the vertebral column from the craniocervical junction to the sacrum (Figure 1). Intracranially, no masses were demonstrable at any of the cranial nerves. No optic or hypothalamic gliomas or meningiomas were demonstrated. Further, it was shown that numerous dumbbell-neurofibromas encroached the spinal canal through the enlarged intervertebral foramina at several cervical and lumbar levels and caused myelum- and cauda-equina compression (Figure 2). These intradural extramedullary tumours led to a series of surgical interventions. Histo-pathologic examination revealed a homogenous morphologic pattern in all spinal neurofibromas composed of spindle-shaped tumour cells in a myxomatous stroma enclosing collagen fibres and ganglion cells. Immunhistochemical study was pathologically positive with the diagnosis of neurofibromas. In addition, one excised cutaneous lesion also was diagnosed as being a neurofibroma.

T2-weighted MRT of the thoracic spine: (a) Neurofibromas (arrows) affecting all spinal nerve roots shown in frontal section. Contrary to the findings in the cervical and in lumbar spine, these neurofibromas extend laterally without involving the spinal canal. (b) This is confirmed by the normal image of the thoracic myelum and the entire midsagittal section.

Contrast-enhanced T1-weighted magnetic resonance tomography (MRT) of the cervical spine: (a) Neurofibromas at the C2-, C3- and C4-spinal nerve roots extending far into the spinal canal (white arrows). (b) Axial section through C3/4 demonstrates the typical enlarged intervertebral foramen caused by neurofibromatous tumour growth (black arrows) and severe myelum compression (black arrowheads). Note that also the contralateral nerve root appears thickened as it is enhanced by contrast uptake (white arrow).
The patient herself reported that NF1 was suspected from the age of 10 years when several CLS were found and four to five lumps in her left leg and over the right muscles. On admittance, patient NF1399 fulfilled the NIH criteria of NF1 due to the presence of numerous cutaneous neurofibromas in addition to CLS and axillary freckling (Table 1).
According to the patient's report both her parents did not show any signs of NF1, such as CLS, freckling or neurofibromas. Neither of her parents had a medical history of brain or spinal cord tumours nor did they show scoliosis or other osseous lesions. Further, patient NF1399 reported that her sister (NF1500) has been diagnosed with NF1, as well, due to three lumps at her left ankle and calve. However, patient NF1500 did not show CLS, freckling, Lisch nodules or other clinical signs of NF1. Neuroimaging did not reveal spinal neurofibromas in the patient NF1500.
Molecular analysis
For protein-truncation testing (PTT) total RNA of the patient NF1399 and of healthy individuals was extracted from PHA-stimulated short-term lymphocyte cultures treated with and without puromycin.12 The entire cDNA of the NF1 gene was PCR amplified in five overlapping fragments using primer pairs as described.13 PCR products were subjected to a transcription/translation reaction and the synthesised polypeptide fragments subjected to electrophoretic separation.
The analysis of the second segment (S2) nucleotides 1486-3583 (exon 10B to 21) of the NF1 cDNA revealed in the patient a shortened fragment in addition to the normal product (Figure 3a). According to the size of the shortened peptides the mutation was suspected within the second half of the analysed fragment. Therefore, a RT–PCR product of exons 15–21 of the NF1 gene was generated from RNA of the patient using the sense primer P714 and the antisense primer of fragment S2. The PCR product was cloned into an AT-cloning vector and individual clones were subjected to sequence analysis. Loss of exon 20 was detected in five of 14 analysed clones. In one clone the first 39 bp of exon 20 were missing due to the use of a cryptic splice acceptor site present in exon 20. The remaining eight clones contained full-length cDNA. These results were highly indicative for a mutation affecting the splice acceptor site of exon 20. For sequence analysis of exon 20 and its flanking intronic sequences genomic DNA was digested with the restriction enzyme BmyI, that cuts specifically the NF1 pseudogene DNA sequence mapped to chromosome 15 (accession no. AC023191 or AC037471), but leaves the sequence of the NF1 gene itself uncut. Subsequently, PCR amplification was performed using primers specific for exon 20 as described.15 We identified a C to G transversion at the base-3 of the 5′-splice site of exon 20 in the patient NF1399 (Figure 3b). Sequence analysis of exon 20 clearly excluded the presence of this mutation IVS19b-3C>G in the patient's sister NF1500, in whom also PTT was unable to reveal any other truncating mutation. Mutation IVS19b-3C>G has been described previously in a classic NF1 patient.16

(a) PTT results of segment S2 of a control person (C) and our patient (pat.). RNA for RT–PCR was extracted from PHA stimulated short term cultivated blood lymphocytes treated with (+P) and without puromycin (−P). The wild-type (wt) and aberrant band caused by loss of exon 20 (3314del182) are indicated. (b) Sequencing results of exon 20 and its adjacent intronic sequences of the patient and a control individual (contr.). In the patient heterozygosity for C and G at base-3 of the splice acceptor site of intron 19b is indicated by an arrowhead.
Discussion
We present here a NF1 patient with exceptionally severe spinal involvement. Spinal neurofibromas, which frequently affect multiple vertebral levels, are found in up to 38% of NF1 patients.4,6,7,8 However, they cause clinical implications only in about 5% of the patients.3,4,5 In contrast, multiple symptomatic spinal neurofibromas are the main clinical finding in patients with familial spinal neurofibromatosis.7,17,18,19 Because of the extensive phenotypic variability typical of NF1 even among members of the same family, the observation of spinal neurofibromatosis in all affected members is striking. Thus, familial spinal neurofibromatosis FSNF or SNF has been regarded as an alternate form of NF1. In addition to the presence of multiple spinal neurofibromas FSNF is characterised by very mild cutaneous signs of NF1 in the affected patients. Based on three multi generation families CLS were considered the sole cutaneous sign present in all affected family members.7,17,19 Recently, Kaufmann et al18 described two families which presented with severe spinal involvement, but did not show CLS. Although overall the cutaneous features are very mild in the affected in these families several of them show additional dermal features as may be deduced from Table 2. Thus, several of the probands in these families fulfilled the criteria for classic NF1 if the cutaneous signs were taken into account.
Due to the lack of other affected family members the diagnosis of FSNF may not unequivocally be made in the here presented index patient. Further, our patient does not fit the strict criteria for SNF, since she shows cutaneous features that are sufficient to fulfill the NIH criteria for NF1. However, the severe spinal involvement in this patient is striking and may indicate that this patient belongs to the distinct clinical entity of spinal neurofibromatosis.
It has been discussed that the type of NF1 mutation may have a specific effect that explains the severe neurological phenotype of SNF.17 The splice mutation found in our patient has also been identified in a classic NF1 patient and her similarly affected father.16 Spinal MRI of the latter patient did not reveal any evidence of spinal neurofibromas, hence demonstrating a lack of genotype-phenotype correlation with respect to this specific mutation. The clinical phenotypes of the patients carrying the identical mutation are summarised in Table 1. Similarly, Kaufmann et al18 found in one of the described families with severe spinal involvement a recurrent splice mutation that caused classical NF1 in another unrelated patient. Furthermore, it is assumed that most germline mutations in NF1 patients result in haploinsufficiency through the reduction of neurofibromin levels.13,15,20 Hence, the involvement of modifying genes is currently the most likely explanation for the clinical phenotype of spinal neurofibromatosis.
The here-presented NF1 case further illustrates the difficulties of counselling patients with border-line clinical features of NF1. This is of particular interest in cases of suspected spinal neurofibromatosis which is hallmarked by very mild cutaneous expression. Moreover, oligosymptomatic expression of NF1 has been reported also in patients without spinal involvement.21 Our index patient's sister (NF1500) has been suspected to have NF1 for many years due to her first degree relationship with a NF1 patient and multiple lumps at her calves that were in location and size comparable to her sister's (NF1399) neurofibromas. However, no other clinical signs of NF1 were detectable and neuroimaging in patient NF1500 did not reveal spinal neurofibromas. Moreover, the medical history of the parents who were not available for further clinical or molecular investigation was unremarkable. Thus, molecular analysis rendered the clinical diagnosis for NF1 in patient NF1500 highly ambiguous.
The case of the here-described sisters demonstrates that reaching conclusions on the genotype-phenotype correlation in NF1 remains a major challenge. The latter may be met adequately only if the phenotypic descriptions of the patients are carefully detailed on the one hand and if more powerful mutation detection technologies16 are applied on the other hand.
References
Easton D, Ponder M, Huson S, Ponder B . An analysis of variation in expression of neurofibromatosis (NF) type 1 (NF1): evidence for modifying genes Am J Hum Genet 1993 53: 305–313
Tonsgard J, Yelavarthi K, Cushner S, Short M, Lindgren V . Do NF1 gene deletions result in a characteristic phenotype? Am J Med Genet 1997 73: 80–86
von Deimling A, Krone W, Menon A . Neurofibromatosis type 1: pathology, clinical features and molecular genetics Brain Pathol 1995 5: 153–162
Tonsgard JH, Kwak SM, Short MP, Dachman AH . CT imaging in adults with neurofibromatosis-1: frequent asymptomatic plexiform lesions Neurology 1998 50: 1755–1760
Huson S, Harper P, Compston D . Von Recklinghausen neurofibromatosis. A clinical and population study in south-east Wales Brain 1988 111: 1355–1381
Thakkar S, Feigen U, Mautner V . Spinal tumours in neurofibromatosis type 1: an MRI study of frequency multiplicity and variety Neuroradiology 1999 41: 625–629
Pöyhönen M, Leisti E, Kytola S, Leisti J . Hereditary spinal neurofibromatosis: a rare form of NF1? J Med Genet 1997 34: 184–187
Egelhoff J, Bates D, Ross J, Rothner A, Cohen B . Spinal MR findings in neurofibromatosis types 1 and 2 Am J Neuroradio 1992 13: 1071–1077
Carey J, Viskochil D . Neurofibromatosis type 1: A model condition for the study of the molecular basis of variable expressivity in human disorders Am J Med Genet 1999 26: 7–13
Viskochil D, Carey J . Alternate and related forms of the neurofibromatoses in Huson S, Hughes R (eds) The neurofibromatoses: a clinical and pathogenetic overview London 1994 pp 445–474
Riccardi V . Neurofibromatosis: phenotype, natural history, and pathogenesis 2nd. edition Baltimore: Johns Hopkins University Press 1992
Wimmer K, Eckart M, Rehder H, Fonatsch C . Illegitimate splicing of the NF1 gene in healthy individuals mimics mutation-induced splicing alterations in NF1 patients Hum Genet 2000 106: 311–313
Heim RA, Kam-Morgan LNW, Binnie CG, Corns DD, Cayouette MC, Farber RA, Aylsworth AS, Silverman LM, Luce MC . Distribution of 13 truncating mutations in the neurofibromatosis 1 gene Hum Mol Genet 1995 4: 975–981
Eisenbarth I, Beyer K, Krone W, Assum G . Toward a survey of somatic mutations of the NF1 gene in benign neurofibromas of patients with neurofibromatosis type 1 Am J Hum Genet 2000 66: 393–401
Fahsold R, Hoffmeyer S, Mischung C et al. Minor lesion mutational spectrum of the entire NF1 gene does not explain its high mutability but points to a functional domain upstream of the GAP-related domain Am J Hum Genet 2000 66: 790–818
Messiaen L, Callens T, Mortier G et al. Exhaustive mutation analysis of the NF1 gene allows identification of 95% of mutations and reveals a high frequency of unusual splicing defects Hum Mut 2000 15: 541–555
Ars E, Kruyer H, Gaona A et al. A clinical variant of neurofibromatosis type 1: familial spinal neurofibromatosis with a frameshift mutation in the NF1 gene Am J Hum Genet 1998 62: 834–841
Kaufmann D, Müller R, Bartelt B et al. Spinal neurofibromatosis without café-au-lait macules in two families with null mutations of the NF1 gene Am J Hum Genet 2001 69: 1395–1400
Pulst S, Riccardi V, Fain P, Korenberg J . Familial spinal neurofibromatosis: clinical and DNA linkage analysis Neurology 1991 41: 1923–1927
Hoffmeyer S, Assum G, Griesser J, Kaufmann D, Nurnberg P, Krone W . On unequal allelic expression of the neurofibromin gene in neurofibromatosis type 1 Hum Mol Genet 1995 4: 1267–1272
Buske A, Gewies A, Lehmann R et al. Recurrent NF1 gene mutation in a patient with oligosymptomatic neurofibromatosis type 1 (NF1) Am J Med Genet 1999 86: 328–330
Acknowledgements
We would like to thank the patients who participated in this study. In particular we wish to thank Ms Martina Vacik for her enthusiastic co-operation. This work was supported by the Jubiläumsfonds der Österreichischen Nationalbank grant no. 7519 and by Het Bijzonder Onderzoeksfonds UG No. 01107799 (L Messiaen).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Wimmer, K., Mühlbauer, M., Eckart, M. et al. A patient severely affected by spinal neurofibromas carries a recurrent splice site mutation in the NF1 gene. Eur J Hum Genet 10, 334–338 (2002). https://doi.org/10.1038/sj.ejhg.5200807
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.ejhg.5200807
Keywords
This article is cited by
-
Do non-pathogenic variants of DNA mismatch repair genes modify neurofibroma load in neurofibromatosis type 1?
Child's Nervous System (2022)
-
Familial spinal neurofibromatosis due to a multiexonic NF1 gene deletion
neurogenetics (2011)
-
The spectrum of somatic and germline NF1 mutations in NF1 patients with spinal neurofibromas
neurogenetics (2009)
