
A package of papers investigates the functional regulatory elements in genomes that have been obtained from human tissue samples and cell lines. The implications of the project are presented here from three viewpoints. See Articles p.317, p.331, p.337 & p.344 and Letters p.350, p.355, p.360 & p.365
The topic in brief
-
Epigenomics is the study of the key functional elements that regulate gene expression in a cell.
-
Epigenomes provide information about the patterns in which structures such as methyl groups tag DNA and histones (the proteins around which DNA is packaged to form chromatin), and about interactions between distant sections of chromatin.
-
They also contain information about regulatory elements in DNA itself: both those that lie in the promoter region immediately upstream of where a gene's transcription begins, and those in distant enhancer sequences.
-
The ENCODE Project1 aimed to catalogue the regulatory elements in human cells, studying the epigenomic signatures of cells grown in culture. The Roadmap Epigenomics Project2,3,4,5,6,7,8,9 builds on this by analysing samples taken directly from human tissues and cells — embryonic and adult, diseased and healthy (Fig. 1).
Figure 1: From body to bench. 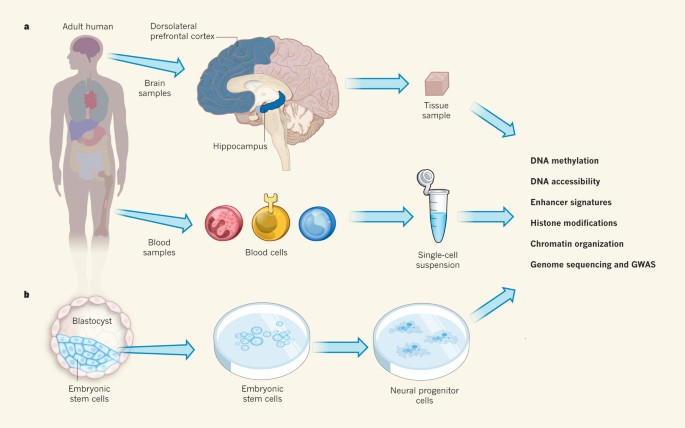
The Roadmap Epigenomics Project has produced reference epigenomes that provide information on key functional elements controlling gene expression in 127 human tissues and cell types2,3,4,5,6,7,8,9, and encompassing embryonic and adult tissues, from healthy individuals and those with disease. a, Many of the adult tissues investigated were broken down by cell type or region — blood into several types of immune cell, for instance, and the brain into regions including the hippocampus and dorsolateral prefrontal cortex. Tissue samples and cells were subjected to a range of epigenomic analyses, along with genome sequencing and genome-wide association studies (GWAS). b, Embryonic stem (ES) cells, which are taken from the embryo at the 'blastocyst' stage and can give rise to almost every cell type in the body, were used to analyse, for example, the differentiation of stem cells into different neuronal lineages. The ES-cell-derived cell lines underwent the same epigenomic analyses as the tissue samples.
-
The researchers have linked these epigenomic data to the corresponding genetic information, producing reference epigenomes for 127 tissue and cell types.
-
The result is a representation of how epigenomic elements regulate gene expression in the human body.

Differentiation enhanced
Casey E. Romanoski & Christopher K. Glass
All the cells in the body contain essentially the same genome, and arise from the progeny of a single fertilized egg. How does each cell type interpret this common set of instructions to achieve its specific identity? The Roadmap Epigenomics Project has tackled this question by defining the epigenomic signatures of a broad spectrum of human tissues and cells undergoing crucial developmental transitions (for an overview2, see page 317). Collectively, these papers and the associated data sets provide an unprecedented resource for understanding relationships between cells and tissues, and for delineating how cell-specific programs of gene expression are achieved.
Only about half of the approximately 25,000 protein-coding genes that make up the mammalian genome are expressed in any given cell type. Although many of these genes are required for general functions and are ubiquitously expressed, others are active in only one or a few cell types, or exhibit different patterns of regulation from cell to cell. A remarkable achievement of the ENCODE Project was the use of epigenomic signatures to infer the existence of hundreds of thousands of enhancer-like regions in the mammalian genome that regulate gene expression at long range. From this vast palate, each cell type is regulated by a subset of perhaps 20,000–40,000 enhancers, which determine its particular gene-expression profile.
Enhancers are activated through interactions with transcription factors, which recognize and bind to specific DNA sequences within the enhancer region. Bound transcription factors recruit co-regulators, many of which deposit or remove modifications on histones. The way in which each cell type interprets genomic information is therefore closely linked to the organization of its DNA regulatory elements. Enhancers that are active in cell-type-specific epigenomic signatures are typically highly enriched in DNA sequences to which lineage-determining and signal-dependent transcription factors bind. Therefore, the delineation of a particular cell's active enhancer repertoire provides a powerful means of predicting the transcription factors required for that cell's identity. By extension, changes in epigenomic signatures during developmental transitions reflect activation or inhibition of such factors.
Four of the papers in this issue2,3,4,5 exploit these relationships to identify combinations of transcription factors that might define different cell types during development. Ziller et al.4 (page 355) modelled neuronal development in vitro, by generating six lineages of neuronal progenitors from embryonic stem (ES) cells, which give rise to almost every cell type of the body. The authors developed computational models to predict the transcription factors that bind to core neural-differentiation enhancers, as well as those that bind enhancers of distinct neural lineages only.
Tsankov et al.5 (page 344) studied the sets of transcription factors that bind to promoters and enhancers in the first three cell lineages that differentiate from ES cells. Sequences bound by transcription factors in one of the three lineages exhibited molecular modifications that promote gene expression, such as loss of DNA methylation. By contrast, the same DNA regions exhibited repressive modifications in the other two cell types. Both Ziller et al. and Tsankov et al. found that regulatory elements controlling genes that are essential for cellular identity are often also epigenetically modified in parental cells, highlighting the importance of existing regulatory landscapes and stage-specific expression of transcription factors for defining the developmental potential of cells.

Some major caveats should be noted. These studies are based on analysis of cell populations, and therefore miss potentially crucial aspects of cellular variability within populations. When tissues are examined, enhancer landscapes represent the composite of the cell types that make up that tissue, not a pure cell population. Studies10,11 of different populations of white blood cells called macrophages suggest that the tissue environment can shape enhancer landscapes, emphasizing the value of studying purified cell populations from in vivo sources. Finally, although the DNA sequences found in cell-specific enhancers provides clues to the identities of the transcription factors that regulate enhancer activation, functional roles must be validated experimentally. The Roadmap Epigenomics Project has made some efforts along these lines, but the large number of hypotheses generated by the current papers means that this step is largely left for future work.
Diseases mapped
Hendrik G. Stunnenberg
For decades, biomedical science has focused on ways of identifying the genes that contribute to a particular trait, or phenotype. Approaches such as genome-wide association studies12 (GWAS) identify locations in the human genome at which variations in DNA sequence are linked to specific phenotypes, but if the variant is located in a region of DNA that does not encode a protein, such studies rarely provide insights into the regulatory mechanisms underlying the association. In these cases, comprehensive epigenomic analyses can provide the missing link between genomic variation and cellular phenotype.
The various consortia, including the Roadmap Epigenomics Program, that are gathered under the umbrella of the International Human Epigenome Consortium (www.ihec-epigenomes.org) have taken up the challenge of deciphering hundreds of cell-type-specific epigenomes using human cells and tissues from healthy donors and people with disease. In this issue, the Roadmap Epigenomics Project presents a wealth of epigenomes, a resource that provides a plethora of new hypotheses to be tested in relation to human health and disease. Given that epigenomes are cell-type specific, it makes sense to analyse disease-associated variants identified by GWAS in the context of the epigenome of the disease cell type. Indeed, previous groundbreaking observations13 revealed that non-protein-coding genetic variants that are associated with phenotypic changes are often located in tissue-specific regulatory regions. The current papers use innovative analytical approaches to deepen and extend this knowledge.
Gjoneska et al.6 (page 365) made use of a mouse model of neurodegeneration that mimics Alzheimer's disease. They found that disease-related changes in gene expression in the hippocampus of the mouse brain correlate with those in post-mortem brain samples taken from people with Alzheimer's disease, but not with those from people without the disease. Subsequent detailed analyses revealed an upregulation of genes and regulatory regions linked to immune responses seen in Alzheimer's disease. Genetic variants associated with the condition seemed to be enriched within evolutionarily conserved regulatory elements that control immune pathways, but not in neuronal pathways, providing fresh entry points for treatment.
Farh et al.7 (page 337) developed an algorithm to identify non-protein-coding genetic variants that might underlie autoimmune disease. The authors found that these variants are often located in or near enhancers or promoters. However, only a small fraction of the variants cause a change in a sequence at which transcription factors are known to bind. This suggests that there is more to an enhancer than a 'simple' collection of sites of transcription-factor binding embedded in the composition of its DNA sequence. For example, flanking sequences might have a topological role affecting chromatin packaging and, consequently, DNA accessibility.
Polak et al.8 (page 360) investigated the distribution of cancer-associated genetic mutations in a set of diverse cancers, and correlated them with cell-type-specific epigenomic features. They found that the mutation profile of each cancer could often be predicted from the epigenomic signature of the cell type from which that cancer was most likely to have originated. Remarkably, the epigenomic signatures of cancer-cell lines (which are often used to study disease) were poor predictors of this profile. The authors conclude that the density and distribution of cancer mutations are strongly linked to a cell-type-specific epigenomic signature.
What comes next? The Roadmap Epigenomics Project has reached a major milestone, but the epigenomes of 127 cell types are just the beginning of the road to a comprehensive epigenome encyclopaedia. The International Human Epigenome Consortium plans to determine the epigenomes of every cell type in the human body — estimated to be several hundred to a thousand. Furthermore, each cell type must be analysed in many individuals, to assess the effect of genetic variation on personal cell-type-specific epigenomes. Finally, studies monitoring the epigenomic changes that arise as a result of ageing and of changes in environmental factors such as nutrients and metabolites will also be interesting. The epigenomics project has taught us that analysis and comparison of the genome and epigenome of healthy and diseased cells is essential for detecting and understanding the drivers of multifactorial diseases and traits.
Chromatin charted
Laurence Wilson & Genevieve Almouzni
Chromatin is the complex of DNA, RNA and proteins that packages DNA within the cell. At the core of chromatin is an eight-subunit protein complex composed of histones. Molecular modifications to either DNA or histones can affect the structure and function of chromatin. For example, some modifications promote chromatin compaction, affecting how easily DNA can be accessed by transcription factors, whereas others act as signals that modulate gene expression. A case in point is modification of the amino-acid residue lysine 27 (K27) on histone H3 in chromatin. Addition of an acetyl group (a modification known as H3K27ac) correlates with transcription of the corresponding region of DNA, whereas trimethylation (H3K27me3) is linked to transcriptional repression.
Several papers published by the Roadmap Epigenomics Project investigate histone modifications, and provide insights into the relationship between histone signatures and gene expression throughout development and adult life. For instance, three studies investigate the histone modifications associated with disease6,7,8. Focusing on normal development, Tsankov et al.4 and Ziller et al.5 have mapped histone modifications that occur during the differentiation of embryonic cells (specifically, H3K4me1, H3K4me3, H3K27ac and H3K37me modifications), alongside patterns of transcription-factor binding and DNA methylation. They describe chromatin remodelling events that alter the accessibility of DNA sequences to which combinations of key regulatory transcription factors bind. These events correlate with the changes in gene expression that occur as cells differentiate.
In addition to the linear viewpoint of chromatin alterations presented through histone modifications, long-range chromatin interactions can also modulate gene expression — for instance, by bringing distant enhancers into contact with promoters that regulate the same gene. Dixon et al.9 (page 331) investigated this phenomenon, charting changes in three-dimensional (3D) chromatin organization during stem-cell differentiation. Human cells contain two copies, or alleles, of each gene, which can vary in terms of DNA sequence, resulting in differences in transcriptional activity (allele-restricted transcription). The allelic complement of a cell is known as its haplotype. Strikingly, Dixon and colleagues report that different haplotypes display different histone modifications and 3D chromatin organization, correlating with its allele-restricted transcription.
Leung et al.3 (page 350) confirmed this observation, reporting haplotype-specific differences in histone modifications and chromatin architecture that correlate with allele-restricted transcription across many tissues. Notably, these differences also correlate with mutations that disrupt sites of either transcription-factor binding or long-range chromatin interactions. However, the functional relevance of these imbalances remains to be deciphered.
These eight studies showcase the use of the first large-scale reference epigenome database, taking advantage of the statistical power afforded by large sample sizes to formulate hypotheses about the relationships between the epigenome and the genome in different biological processes. They strengthen the link between chromatin modifications and gene expression in development and disease, defining core regulatory circuits that act in different tissues and at different developmental stages. This provides the community with a powerful reference tool, allowing researchers to compare the epigenome in their tissue of choice with snapshots from the database.
It is, however, still early days. Future work should try to address the changing relationship between the epigenome and genome over the lifespan of the cell, in different phases of the cell cycle and across cellular generations. Other factors that modulate chromatin organization also remain to be investigated — the proteins responsible for chromatin remodelling, for example, and the chaperone proteins associated with histone variants that control assembly and disassembly of chromatin14.
Defining the mechanisms that underlie chromatin-based regulation of gene expression will require integration of the observations made by the Roadmap Epigenomics Project with other approaches that directly test for function. For instance, model organisms will remain essential for comparative epigenomics and for garnering evolutionary information. Cutting-edge techniques, such as high-resolution microscopy, will allow live imaging of chromatin architecture and a means of studying its dynamics in space and time.
Above all, approaches and technologies that draw from different disciplines must be integrated in future epigenomic projects. This multidisciplinary approach is being catalysed by collaborations such as the EpiGeneSys network (www.epigenesys.eu), which bridges epigenetics and systems biology. Combining such efforts will be essential for understanding the functional link between the epigenome and the genome. Footnote 1
Notes
References
The ENCODE Project Consortium. Nature 489, 57–74 (2012).
Roadmap Epigenomics Consortium. Nature 518, 317–330 (2015).
Leung, D. et al. Nature 518, 350–354 (2015).
Ziller, M. J. et al. Nature 518, 355–359 (2015).
Tsankov, A. M. et al. Nature 518, 344–349 (2015).
Gjoneska, E. et al. Nature 518, 365–369 (2015).
Farh, K. K.-H. et al. Nature 518, 337–343 (2015).
Polak, P. et al. Nature 518, 360–364 (2015).
Dixon, J. R. et al. Nature 518, 331–336 (2015).
Gosselin, D. et al. Cell 159, 1327–1340 (2014).
Lavin, Y. et al. Cell 159, 1312–1326 (2014)
Welter, D. et al. Nucleic Acids Res. 42, D1001–D1006 (2014).
Maurano, M. T. et al. Science 337, 1190–1195 (2012).
Gurard-Levin, Z. A., Quivy, J.-P. & Almouzni, G. Annu. Rev. Biochem. 83, 487–517 (2014).
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article

Cite this article
Romanoski, C., Glass, C., Stunnenberg, H. et al. Roadmap for regulation. Nature 518, 314–316 (2015). https://doi.org/10.1038/518314a
Published:
Issue Date:
DOI: https://doi.org/10.1038/518314a
This article is cited by
-
Methamphetamine-induced region-specific transcriptomic and epigenetic changes in the brain of male rats
Communications Biology (2023)
-
Detection and identification of cis-regulatory elements using change-point and classification algorithms
BMC Genomics (2022)
-
Recent innovations and in-depth aspects of post-genome wide association study (Post-GWAS) to understand the genetic basis of complex phenotypes
Heredity (2021)
-
Broad genic repression domains signify enhanced silencing of oncogenes
Nature Communications (2020)
-
Evaluation of post-translational modifications in histone proteins: A review on histone modification defects in developmental and neurological disorders
Journal of Biosciences (2020)
