
Key Points
-
Crystal deposition diseases are a differential diagnosis for preauricular pain, swelling and limited mouth-opening along with the more obvious temporomandibular joint dysfunction or parotitis
-
Look for clues in the medical history such as hyperparathyroidism or joint pain/arthritis
-
Examine the temporomandibular joints on panoral films
-
Refer in for a joint aspirate or to a rheumatologist if suspicious
Abstract
Pseudogout is an acute presentation of one type of crystal deposition disease in which calcium pyrophosphate dihydrate crystals are found in the joint spaces of synovial joints. In this case, a 56-year-old caucasian male presented with right sided preauricular swelling, temporomandibular joint arthralgia and restricted mouth opening; he developed identical symptoms on the left side two days later.
Similar content being viewed by others
Main
Crystal deposition diseases are a collection of arthropathies in which crystals are found in synovial fluid.1 There are three principal types: urate depostion (gout), calcium pyrophosphate dihydrate deposition (pseudogout), and hydroxyapatite deposition (calcific periarthritis). Each crystal deposit is associated with a different clinical syndrome, and each are relatively rare conditions.
Calcium pyrophosphate dihydrate (CPPD) deposition disease is a rare, benign condition in which CPPD crystals are deposited in synovial fluid2 and which results in the calcification of articular cartilage (chondrocalcinosis). This leads to an acute arthritis (or 'pseudogout') in 25% of patients.3 These acute episodes, where CPPD crystals are shed into synovial fluid and provoke an inflammatory reaction, commonly affect the knee and less commonly other large joints such as the wrist. Rarely, smaller synovial joints such as the temporomandibular joints (TMJs) are affected. Whilst this acute presentation mimics gout, chronic features resemble osteoarthritis.4
The aetiology is incompletely understood, but there are familial relationships with several autosomal dominant subtypes.1 Various genetic studies have linked CPPD to chromosomes 5p, 8p and 15.5,6,7,8 There are also associations with systemic diseases such as hypophosphatasia, hypothyroidism and hyperparathyroidism.
The prevalence is unknown as individuals may not exhibit any symptoms, but McCarty remarks that CPPD crystal deposits have been found in around 5% of the population in general.1 In one study involving over 1000 cases,1 males and females were affected in a ratio of 1.5 : 1, the incidence increasing with age; the mean age of presentation was 72 years.
Case report
A 56-year-old man was referred to the Accident and Emergency (A&E) Department by his general medical practitioner. He complained of acute pain and swelling in the right preauricular region and had difficulty opening his mouth. He reported a similar painful incident 15 years previously, which had been attributed to masticatory muscle spasm and which was apparently relieved by analgesics and muscle relaxants.
He was diagnosed by the A&E doctor as having a recurrent episode of facial arthromyalgia and was prescribed paracetamol 1 g, codeine phosphate 60 mg and diazepam 5 mg. Later the same day, he re-presented at A&E because the pain was progressively worsening despite previous treatment, and he was referred to the department of Oral and Maxillofacial Surgery for an opinion.
Direct questionning elicited a past history of clicking and stiffness from both joints. He could report no initiating or relieving factors for his current pain and swelling, apart from aggravation of pain on mouth opening. There was no increase in swelling periprandially, although he was unable to eat at that time because of trismus and pain.
He gave a history of hypertension and angina, for which he took nifedipine and isosorbide mononitrate. He reported blindness in one eye caused by a retinal artery embolism. A parathyroid adenoma had been excised 5 months previously, and he also mentioned that he had nephrocalcinosis and had pain from a knee joint which was ongoing.
On examination there was a diffuse warm swelling in the right preauricular region, which was very tender to palpation. There was limited mouth opening caused by pain. He had a pyrexia of 38.2 °C, with a neutrophil leucocytosis of 14.6x109 (reference range 2.0–7.5), suggestive of infection. Saliva appeared to drain normally from the parotid duct with no pus evident. His dental health appeared fair with no obvious source of infection. Blood cultures were negative.
The differential diagnoses included a parotitis, an acute exacerbation of osteoarthritis, an infective arthritis or internal derangement of the joint. He was prescribed intravenous augmentin 1.2 g, and continued diazepam, paracetamol and codeine.
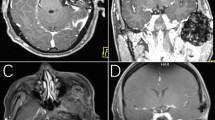
The following day, the symptoms were reducing although he was still pyrexial at 37.3 °C. Radiographic examination (Fig. 1) revealed calcification within both TMJs, with a loss of joint space, flat articular surfaces and periarticular sclerosis; the appearance on the left side was more severe. A provisional diagnosis of an acute inflammatory arthritis was made.

Purely physiological tooth surface loss
After 2 days, the pain had settled and he was apyrexial; however, the left TMJ had become painful, with preauricular swelling, making the diagnosis of acute arthritis more likely. The synovial fluid of the left TMJ was aspirated revealing blood stained fluid and a small amount of yellow granular material, which was sent for microscopy, culture and sensitivity. This revealed rhomboid (needle shaped) crystals on light microscopy, which were weakly positively birefringent under plane polarised light and consistent with the appearance of CPPD crystals. Blood was examined for levels of circulating Rheumatoid Factor, uric acid and HLA-B27, and biochemical analyses including urea and electrolytes, bone biochemistry (calcium and phosphate levels), thyroid function tests and magnesium levels. These were within normal ranges, notably calcium at 2.29 mmol/l (range 2.10–2.70) and phosphate 0.87 mmol/l (range 0.8–1.4).
The final diagnosis was that of calcium pyrophosphate dihydrate crystal deposition disease, with a presentation of pseudogout sequentially in the right, then left TMJ.
The patient was referred to a rheumatologist for further investigation, who also reported chondrocalcinosis to be evident in the patient's knees and shoulders.
Discussion
The first reported case of CPPD was in the early 1900s by Bennett.9 Subsequently it was termed pseudogout because of its similar presentation to gout, although the term is now reserved for an acute episode. Pseudogout manifests as acute pain and swelling of a joint, with heat, effusion and limited mobility; it therefore can resemble osteoarthritis, gout, calcific periarthritis and other inflammatory arthritides and neoplasms. In the case described, the joint affected was a temporomandibular joint, which is rarely affected by crystal deposition diseases. Only one paper has reported CPPD to affect the TMJs bilaterally.10 Acute presentation of CPPD often occurs in the large joints, such as the knee (over 50%)11 or wrist.
Commonly, individuals become aware of CPPD following an episode of painful pseudogout, as happened in this case. They may, however, have a chronic form which resembles osteoarthritis or rheumatoid arthritis in its presentation.
CPPD is diagnosed by light microscopy of synovial fluid aspirate and by radiography of the joint, with haematology and biochemistry to eliminate other arthropathies. The aspirate classically is bloodstained and thick and milky in appearance, as with this patient, caused by the large number of crystals present. Light microscopy demonstrates crystals of calcium pyrophosphate dihydrate which are weakly positively birefringent under polarised light microscopy; these are often present phagocytosed within neutrophils in acutely inflamed joints. The microcrystalline aggregates may measure from 15 micrometres to 6 millimetres in diameter.1
Haematology results often show a neutrophil leucocytosis, as in this case, which may suggest some element of tissue damage; the associated release of interleukin-1 during tissue necrosis explains the accompanying pyrexia. Biochemical analyses may indicate a raised or normal calcium level,2 normal in this case. A raised serum calcium warrants exclusion of raised parathyroid hormone levels. This patient had recently undergone surgery for hyperparathyroidism, which is a known association of CPPD. The erythrocyte sedimentation rate (ESR) may also be raised. Associated tests, not positive in this case, include uric acid levels to confirm or eliminate the presence of gout; and HLA-B27, a histocompatibility antigen found in over 90% of patients with ankylosing spondylitis and over 60% of those with Reiters disease, and is used to eliminate these conditions.
CPPD may present as an incidental finding on a radiograph. A typical radiographic feature is linear calcification of articular cartilage, known as 'chondrocalcinosis articularis'.12 Similar joint changes, consistent with this appearance, were evident on this patient's orthopantomogram. One study13 suggested that this appearance was age related: patients admitted to one geriatric unit had radiographs taken of their hands, knees, wrists and pelvis; this showed radiographic evidence of chondrocalcinosis present in 36% of 75–84-year-olds, and 44% of those over 84 years.
Diagnosis is definite where CPPD crystals are demonstrated by chemical analysis or where crystals compatible with CPPD are demonstrated by light microscopy and there is a typical radiographic appearance. If only one of the previous diagnostic criteria is found, the diagnosis is probable.
The aetiology of this condition may be divided into:1
-
1
Hereditary – often autosomal dominant
-
2
Sporadic (idiopathic)
-
3
Those cases associated with metabolic disease, for example hyperparathyroidism
-
4
Those cases associated with trauma or surgery
Research into finding the gene locus for this condition, particularly in inherited and sporadic forms of this disease, has been carried out most notably over the past 5 years. A study carried out in Europe in 1995 linked a familial form of the condition to chromosome 5p,5 and further work carried out linked it more closely to 5p15, the short arm of chromosome 5.6,7 Another team has linked it to chromosome 8p as part of a syndrome with an early onset osteoarthritis,8 and it has recently been associated with chromosome 15.14
The pathogenesis of calcium pyrophosphate dihydrate crystal deposition is not fully understood. Accepted theory1 relates CPPD crystal deposition to a disturbance in calcium or inorganic phosphate metabolism, leading to an excess of either in joints. For example, serum calcium changes acutely following parathyroidectomy, which has a well recognized association with pseudogout.2 These elevated levels, with or without changes in the matrix which promote crystal formation, are thought to lead to the formation of calcium pyrophosphate dihydrate crystals in cartilage. This is substantiated by recent research14 on the mouse 'ank' gene, which has implicated a specific gene on chromosome 15 in the pathogenesis of CPPD crystal deposition. This gene codes for a transmembrane protein which regulates pyrophosphate transport (affecting local phosphate metabolism) and tissue calcification. The 'ank' gene has also been linked to the matrix GLA protein which is associated with articular calcification;15 as mentioned, changes in the matrix can promote crystal formation. These mouse mutations are still being researched.15
Pseudogout, the acute presentation of CPPD, is thought to be related to crystals being shed from the cartilage surface – possibly precipitated by damage to cartilage, whether caused by trauma, infection or inflammation. Surgical procedures or severe illness (such as stroke) are also known to provoke an acute attack.
Treatment requires thorough arthrocentesis to remove the crystals, the prescription of anti-inflammatories (steroidal and non steroidal), joint immobilization, the provision of systemic corticosteroids and rarely the use of colchicine. The use of intramuscular triamcinolone acetonide 60 mg was assessed in 14 patients during an acute attack of pseudogout in one study16 and found to give 50% improvement in symptoms. Colchicine is more often associated with the treatment of gout, and is intended to be given during an acute phase. It inhibits the release of a chemotactic factor by polymorphonuclear leucocytes17 after phagocytosis of CPPD crystals.1 0.5–2 milligrams of colchicine intravenously may predictably control acute pseudogout.18 Analgesia and anti-inflammatories are of value but not as effective as in the treatment of gout.3
There is no known way of preventing continual CPPD deposition in joints, and this tends to recur. It is simply advisable to correct existing precipitants such as hyperparathyroidism, and to treat the disease symptomatically.
References
McCarty DJ, Koopman WJ Arthritis and allied conditions – a textbook of rheumatology. 13th ed pp 2103–2126 Baltimore, London: Williams and Wilkins 1997.
Kobayashi S, Sugenoya A, Takahashi S et al. Two cases of acute pseudogout attack following parathyroidectomy. Endocrinol Jpn 1991; 38: 309–314.
Kumar, Clark Clinical Medicine. 3rd ed pp 410–411 London: Balliere Tindall 1994.
Moll J Rheumatology, a colour guide. 3rd ed pp 63 Edinburgh: Churchill Livingstone 1992.
Hughes AE, McGibbon D, Woodward E, Dixey J, Doherty M Localisation of a gene for chondrocalcinosis to chromosome 5p. Hum Mol Genet 1995; 4: 1225–1228.
Andrew LJ, Brancolini V, de la Pena LS et al. Refinement of the chromosome 5p locus for familial calcium pyrophosphate dihydrate deposition disease. Am J Hum Genet 1999; 64: 136–145.
Rojas K, Serano de la Pena L, Gallardo T Physical map and characterization of transcripts in the candidate interval for familial chondrocalcinosis at chromosome 5p15.1. Genomics 1999; 62: 177–183.
Reginato AJ, Tamesis E, Netter P Familial and clinical aspects of calcium pyrophosphate deposition disease. Curr Rheumatol Rep 1999; 1: 112–120.
Bennett EH Abnormal deposits in joints. Dublin J Med Sci 1903; 65: 161–163.
Chuong R, Piper MA Bilateral pseudogout of the temporomandibular joint: report of a case and review of literature. J Oral Max Surg 1995; 53: 691–694.
McCarty DJ, Hogan JM, Gatter RA et al. Studies on pathological calcifications in human cartilage. J Bone Joint Surg Am 1966; 48: 309–325.
Zitnan D, Sitaj S Condrocalcinosis articularis. Section 1. Clinical and Radiological Study. Ann Rheum Dis 1963; 22: 142.
Wilkins Osteoarthritis and articular chondrocalcinosis in the elderly. Ann Rheum Dis 1983; 42: 280–284.
Ho AM, Johnson MD, Kingsley DM Role of the mouse ank gene in control of tissue calcification and arthritis. Science 2000; 289: 265–270.
Maldonado I, Reginato AM, Reginato AJ Familial calcium crystal diseases: what have we learned?. Curr Opin Rheumatol 2001; 13: 225–233.
Roane DW, Harris MD, Carpenter MT et al. Prospective use of intramuscular triamcinolone acetonide in pseudogout. J Rheumatol 1997; 24: 1168–1170.
Zemplenyi J, Calcaterra TC Chondrocalcinosis of the temporomandibular joint. A parotid pseudotumour. Arch Otolaryngol 1985; 3: 403–405.
Spilberg et al. Colchicine and pseudogout. Arthritis Rheum 1971; 14: 109–116.
Acknowledgements
The authors are grateful for the advice of Dr R. K. Jacoby, Consultant Rheumatologist in the management of this case.
Author information
Authors and Affiliations
Corresponding author
Additional information
Refereed paper
Rights and permissions
About this article
Cite this article
Greaves, S., Fordyce, A. Bilateral temporomandibular joint pseudogout. Br Dent J 192, 25–27 (2002). https://doi.org/10.1038/sj.bdj.4801279
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.bdj.4801279
