
Abstract
The chimeric fusion protein AML1-ETO, created by the t(8;21) translocation, recruits histone deacetylase (HDAC) to AML1-dependent promoters, resulting in transcriptional repression of the target genes. We analyzed the transcriptional changes in t(8;21) Kasumi-1 AML cells in response to the HDAC inhibitors, depsipeptide (FK228) and suberoylanilide hydroxamic acid (SAHA), which induced marked growth inhibition and apoptosis. Using cDNA array, annexin A1 (ANXA1) was identified as one of the FK228-induced genes. Induction of ANXA1 mRNA was associated with histone acetylation in ANXA1 promoter and reversal of the HDAC-dependent suppression of C/EBPα by AML1-ETO with direct recruitment of C/EBPα to ANXA1 promoter. This led to increase in the N-terminal cleaved isoform of ANXA1 protein and accumulation of ANXA1 on cell membrane. Neutralization with anti-ANXA1 antibody or gene silencing with ANXA1 siRNA inhibited FK228-induced apoptosis, suggesting that the upregulation of endogenous ANXA1 promotes cell death. FK228-induced ANXA1 expression was associated with massive increase in cell attachment and engulfment of Kasumi-1 cells by human THP-1-derived macrophages, which was completely abrogated with ANXA1 knockdown via siRNA transfection or ANXA1 neutralization. These findings identify a novel mechanism of action of HDAC inhibitors, which induce the expression and externalization of ANXA1 in leukemic cells, which in turn mediates the phagocytic clearance of apoptotic cells by macrophages.
Similar content being viewed by others


Main
The t(8;21) translocation creates the chimeric fusion protein AML1-ETO,1 which recruits the histone deacetylase (HDAC) enzymes to AML1-dependent promoters, resulting in transcriptional repression of the target genes. Histone acetylation is essential for the establishment of transcriptionally competent chromatin.2 The acetylation state of histones and other proteins that regulate transcription is controlled by histone acetyltransferases and histone deacetylases (HDACs).3 HDACs also form a complex with leukemia fusion proteins, such as AML1-ETO, PML-RARα, MLL-CBP, and others, which results in the aberrantly suppressed expression of genes required for the control of cell growth and hence the transformation of primitive hematopoietic cells.4 Conversely, the histone deacetylase inhibitors (HDACIs) are capable of inducing apoptosis, differentiation, and growth in tumor cells, including leukemia cells.5 These properties of HDACIs have made them of particular interest as a means of overcoming the drug resistance characteristic of acute myeloid leukemia (AML) that now reduces the prospects for long-term survival in these patients. Several structurally diverse HDACIs have now been developed as anticancer agents; of these sodium phenylbutyrate, depsipeptide (FK228), suberoylanilide hydroxamic acid (SAHA), and MS-275 are currently being studied in clinical trials in patients with hematologic malignancies and solid tumors.5 The mechanism of HDACIs is unknown, however. Knowledge of their mechanism could be used in the development of other improved HDACIs and in the design of combination treatment.
We therefore studied the mechanisms of the anti-leukemic effects of HDACIs in the AML cell line Kasumi-1. We selected FK228 that was previously shown to possess cytotoxic activity against myeloid leukemia6, 7 but not in T-cell lymphoma cells,8 and used another HDACI SAHA, which is currently undergoing clinical trials in hematologic malignancies.9 To identify genes regulated by HDAC in Kasumi-1 cells harboring the AML1-ETO fusion protein, we first analyzed transcriptional changes in response to FK228 using cDNA array technology. We found that FK228-induced apoptosis was associated with the induction of the expression of the pro-apoptotic protein annexin A1 (ANXA1) through the histone acetylation of its promoter. ANXA1 is a 37 kDa member of the annexin superfamily that structurally belongs to a family of ubiquitous phospholipids and calcium-binding proteins. Depending on the cell system, it is implicated in apoptosis induction,10 caspase-3 activation,11, 12 cell growth inhibition,13, 14 and phagocytosis.15 Further, downregulation of ANXA1 expression has been reported to correlate with tumor progression in head and neck cancer.10 In this study, we discovered that FK228 induced histone acetylation, induction of ANXA1 gene expression and C/EBPα binding in ANXA1 promoter specifically in AML1/ETO-expressing AML cells, and these effects contributed to apoptotic cell death and the engulfment of apoptotic cells by macrophages. These effects were blocked both by the ANXA1-blocking antibody and by silencing of the ANXA1 gene with ANXA1 siRNA. Since the phagocytic clearance of apoptotic leukemia cells plays an important role in the resolution of inflammation after chemotherapy, our findings also emphasize the pivotal role of ANXA1 as a mediator of apoptosis and phagocytosis in response to HDAC inhibition.
Results
FK228 inhibits growth and induces apoptosis of Kasumi-1 cells
We first examined the dose-dependent inhibitory effects of FK228 on the growth of Kasumi-1 cells exposed to incremental concentrations of FK228 (1, 5, and 10 nM) for 16 h. Cell counts were determined at 24, 48, 72, and 96 h after wash-out of the drug (Figure 1a, i). Since treatment with 5 nM FK228 significantly inhibited the growth of Kasumi-1 cells compared with the growth of control cells, this concentration was chosen for further mechanistic studies.


FK228 effects on growth inhibition, apoptosis, and cell cycle in Kasumi-1 and NB4 cells. (a), (c) Kasumi-1 cells (a) and NB4 cells (c) were cultured in the presence of different concentrations of FK228 (1, 5, and 10 nM) for 16 h, followed by wash-out of the drug with medium change. The effects on cell growth (i) were determined by viable cell counts using the Trypan blue exclusion method. Graphs show the mean±S.D. of results of three independent experiments. (ii) Percentage of annexin V-positive control cells and cells treated with FK228 (72 h time point). Graphs show the mean±S.D. of results from three independent experiments. Statistically significant differences (**P<0.01) were determined by a paired Student's t-test. (b) Example of results from flow cytometric cell cycle analysis. Kasumi-1 cells were treated with FK228 (5 nM) for 16 h, followed by wash-out of the drug, after which fixed cells were stained with PI at the 72 h time point. Numbers indicate the average±S.D. of the percentage of cells in the G1 and S phases of the cell cycle from three independent experiments, as calculated by the ModFit program. (d), (e) Kasumi-1 cells (d) and NB4 cells (e) were cultured in the presence of different concentrations of SAHA (0.5, 1.0, and 2.0 μM) for 16 h. The effects on cell growth (i) were determined by viable cell counts using Trypan blue exclusion method. Graphs show the mean±S.D. of results of three independent experiments. (ii), percentage of annexin V-positive control cells and cells treated with SAHA (72 h time point). Graphs show the mean±S.D. of results from three independent experiments. Statistically significant differences (**P<0.01) were determined by a paired Student's t-test
As FK228 has been reported to induce apoptosis in many tumor cell types, we next investigated whether FK228 induced apoptosis in Kasumi-1 cells. At 72 h of culture, the transient (16 h) exposure of cells to 5 nM FK228 induced apoptosis (P<0.001, Figure 1a, ii). At a concentration of 10 nM, FK228 induced the death of almost all Kasumi-1 cells. FK228 at 5 nM also affected the cell cycle, in that the ratio of G0/G1 phase cells moderately increased and the ratio of S-phase cells significantly decreased compared with the findings in untreated control cells at 72 h (P=0.03), with increase in the hypodiploid (sub-G1) population (Figure 1b). Overall, this showed that the FK228-induced cell growth inhibition of Kasumi-1 cells is caused by the combination of apoptotic and cell cycle-inhibitory responses.
As shown in Figure 1c, however, NB4 APL cells, which express fusion protein PML-RARα4 known to form complexes with HDACs, were less sensitive than Kasumi-1 cells to FK228. Although the dose-dependent growth-inhibitory effects of FK228 were likewise observed in NB4 cells, 5 nM FK228 did not induce significant apoptosis and only minimally affected the G0/G1 cell cycle distribution (G0/G1: control 44.8±4.2 versus FK228 57.6±4.0, P=0.04; S: control 49.0±4.2 versus FK228 38.5±2.5, P=0.03, at 72 h). A higher dose (10 nM) of FK228 did, however, significantly inhibit NB4 cell growth and apoptosis, concordant with the findings from a previous study.7
Another HDACI, SAHA, caused the dose-dependent cell growth inhibitory effects and induction of apoptosis in both, Kasumi-1 and NB4 cells (Figure 1d and e). Whereas lower dose of SAHA (0.5 and 1 μM) induced more apoptosis in Kasumi-1 than NB4 cells, higher dose (1 μM) SAHA induced almost complete cell death of Kasumi-1 and NB4 cells.
FK228 upregulates ANXA1 expression in AML1-ETO-positive Kasumi-1 and SKNO-1 cells
To determine the mechanisms of apoptosis induction by FK228, we examined changes in the gene expression of FK228-treated Kasumi-1 cells using a cDNA array. The following criteria were used to identify the genes differentially expressed in FK228-treated and control cells: a lower bound of fold changes (FC) >2 and a difference of means of >150 were used to identify upregulated genes and an upper bound of FC ⩽2 and difference of mean of ⩽150 were used to identify downregulated genes. In FK228-treated Kasumi-1 cells, 123 genes were upregulated and 14 genes were downregulated. Among these, the gene that encodes the tumor necrosis factor α (TNFα) was moderately upregulated (2.1-fold), consistent with a previous report of TNFα gene induction through histone hyperacetylation.16 The anti-inflammatory cytokine TGF-β17 gene was similarly found to be upregulated by 2.3-fold in the FK228-treated Kasumi-1 cells. The genes upregulated >2.1-fold are summarized in Table 1. We focused our studies on the pro-apoptotic ANXA1 gene, which showed a 3.5-fold increase in expression in response to FK228.
To confirm the microarray results, we analyzed changes in the mRNA expression of the ANXA1 gene using real-time PCR. This showed that the ANXA1 mRNA expression in Kasumi-1 cells was repeatedly upregulated by 5 nM FK228 or 1 μM SAHA (FK228 6.4±0.2, SAHA 7.4±0.5 fold increase compared with control; Figure 2a). However, no further increase in the ANXA1 mRNA levels was observed at a higher (10 nM) dose of FK228 (data not shown). Likewise, these HDACIs induced ANXA1 mRNA in another AML1-ETO-positive cell line, SKNO-1 cells (FK228 6.0±0.7-, SAHA 9.6±1.3-fold increase compared with control), but not in the AML1-ETO-negative NB4, U937, or OCI/AML2 cells (Figure 2a). No further induction of ANXA1 mRNA was observed in NB4 cells at a higher dose of FK228 (10 nM) or at later time points (48 and 72 h; data not shown).


FK228 and SAHA induce ANXA1 mRNA with increases in histone acetylation in the ANXA1 promoter in Kasumi-1 and SKNO-1 cells. (a) Induction of ANXA1 mRNA expression in Kasumi-1, SKNO-1, NB4, U937, and OCI/AML2 cells treated with FK228 (5nM), SAHA (1 μM), and/or Z-DEVD-fmk (50 μM) for 24 h, as detected by TaqMan RT-PCR analysis. The abundance of transcripts of ANXA1 relative to the abundance of transcripts of β2-microglobulin was determined as described in the Materials and Methods. Graphs show the representative data from three independent experiments. (b) Chromatin from Kasumi-1, SKNO-1, or NB4 cells treated with FK228 (5 nM) or SAHA (1 μM) for 24 h was immunoprecipitated with an antibody against acetylated H3-Lys9(i) and H4(ii) and then analyzed by TaqMan PCR using probes and primers corresponding to the ANXA1 promoter. The graph shows the abundance of acetylated H3-Lys9 and H4 relative to that of ‘input’ as described in the Materials and Methods. Graphs show the representative data from three independent experiments. (c) Real-time TaqMan PCR profiles of ANXA1 promoter amplification shown by examples of ChIP assays. Representative profiles of acetylated H3-Lys9 in Kasumi-1 (i) and NB4 (ii) cells treated with FK228. (iii) The In put curve shows the total genomic DNA before immunoprecipitation
FK228 activates ANXA1 gene transcription through histone acetylation in AML1-ETO-positive Kasumi-1 and SKNO-1 cells
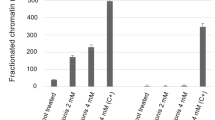
We next determined whether induction of ANXA1 transcription is mediated by histone acetylation. This was examined using the ChIP assay and quantified by TaqMan PCR to identify the critical histone residue on the ANXA1 promoter modified by HDACIs in AML1-ETO-positive and -negative cells. As shown in Figure 2b and c, both FK228 and SAHA promoted histone acetylation in the ANXA1 promoter in AML1-ETO-positive Kasumi-1 cells (histone H3L9; FK228 38.1±7.6-, SAHA 83.6±13.5-, H4; FK228 3.9±0.8-, SAHA 4.1±0.8-fold increase compared with control). Similar results were observed in SKNO-1 cells (H3L9; FK228 3.4±0.6-, SAHA 58.1±9.5-, H4; FK228 1.4±0.3-, SAHA 2.6±0.5-fold increase compared with control) but not in the AML1-ETO-negative cells (i.e., NB4, U937, and OCI/AML2 cells).
To clarify the mechanisms of histone acetylation in the ANXA1 promoter in AML1-ETO-positive cells, we examined the possibility of ANXA1 being a direct target of the AML1-ETO gene. However, no direct AML1-binding sites were identified in the ANXA1 promoter. We next focused on the ANXA1 promoter region as a possible location of binding sites for the candidate AML1 target genes. A survey of this region showed that these sites are repressed by AML1-ETO via HDAC recruitment,18 and identified four candidate C/EBPα (transcription factor CCAAT/enhancer binding protein α) binding sites: tgtgaTTCCAact (GenBank U25414; nucleotides 410–422), aaaTGTAAtattt (nucleotides 610–622), cttTGTAAtgcca (nucleotides 822–834), and gtgTGAAAtcttc (nucleotides 1001–1013). Because expression of the C/EBPα gene and C/EBPα-dependent transcription is inhibited by AML1-ETO through the suppression of its positive autoregulatory loop,19, 20 we investigated whether HDACIs might induce C/EBPα gene expression in AML1-ETO-positive cells. This showed that FK228 or SAHA specifically increased C/EBPα mRNA levels in Kasumi-1 cells (FK228 9.1±0.9, SAHA 4.6±0.5-fold increase compared with control) and SKNO-1 cells (FK228 5.2±0.6-, SAHA 3.3±0.4-fold increase compared with control) but not in NB4, U937, and OCI/AML2 cells (Figure 3a). Using a ChIP assay, we confirmed the recruitment of C/EBPα to the ANXA1 promoter region in Kasumi-1 cells (12.2±3.3-fold increase) and SKNO-1 cells (induction of C/EBPα from no detection in control) following exposure to FK228 (Figure 3b). These findings strongly indicate that the transcription of ANXA1 is regulated by reversal of the HDAC-dependent suppression of C/EBPα by AML1-ETO and the direct recruitment of C/EBPα to the ANXA1 gene promoter.

FK228 and SAHA induce C/EBPα mRNA with increases in C/EBPα recruitment to the ANXA1 promoter in Kasumi-1 and SKNO-1 cells. (a) Induction of C/EBPα mRNA expression in Kasumi-1, SKNO-1, NB4, U937, and OCI/AML2 cells treated with FK228 (5 nM) or SAHA (1 μM) for 24 h detected by TaqMan RT-PCR analysis. The abundance of transcripts of C/EBPα relative to that of β2-microglobulin was determined as described in the Materials and Methods. Graphs show the representative data from two independent experiments. (b) Chromatin from Kasumi-1 and SKNO-1 cells treated with FK228 (5 nM) for 24 h was immunoprecipitated with an antibody against C/EBPα and analyzed by TaqMan PCR using probes and primers corresponding to the ANXA1 promoter. The graph shows the abundance of C/EBPα recruitment relative to that of ‘input’ as described in Materials and Methods. Graphs show the representative data from two independent experiments
FK228-induced ANXA1 promotes apoptosis
We next focused on the investigation of the functional role of ANXA1 in the cellular effects induced by HDAC inhibitors in Kasumi-1 cells. The overexpressions of both, full-length ANXA1 and of the N-terminal cleavage of ANXA1 have been shown to promote apoptosis.11, 12 Immunofluorescence analysis using either monoclonal ANXA1 antibody, which detects the full-length ANXA1, or polyclonal ANXA1 antibody, detecting cleaved N-terminal-truncated protein ANXA1 with full length form, demonstrated increase in ANXA1 expression localized on the plasma membrane in FK228-treated Kasumi-1 cells, more prominently detectable by polyclonal anti-ANXA1 (Figure 4a). The caspase-induced cleaved product of ANXA1 protein has been reported to be associated with cell death,11 and as shown in Figure 4b, we indeed confirmed the increase in the amount of cleaved N-terminal-truncated ANXA1 protein (33 kDa) by FK228 using Western blot analysis with polyclonal anti-ANXA1. Monoclonal anti-ANXA1 antibody also detected the full-length (37 kDa) ANXA1 protein increase by FK228. FK228 initiated caspase-3 cleavage associated with appearance of the increased cleaved version of ANXA1 in Kasumi-1 cells (Figure 4b, ii). Caspase-3 inhibitor Z-DEVD fmk effectively blocked caspase-3 cleavage (Figure 4b, ii), diminished FK228-induced apoptosis (% AnnexinV+ cells: control 16.1±3.2, FK228 54.4±4.6, FK228/ Z-DEVD 34.4±4.5, P=0.01, at 72 h), suppressed accelerated cleavage of ANXA1 (detected by polyclonal antibody) and partially prevented increase in full-length protein (detected by monoclonal antibody; Figure 4b), with no change in ANXA1 mRNA (Figure 2a). These results indicate that the caspase-3 activation in part contributes to the ANXA1 activation and cell death, but these effects are transcription-independent.

(a) ANXA1 induction and translocation induced by FK228 in Kasumi-1 cells. Induction and translocation of ANXA1 in Kasumi-1 cells detected by immunofluorescence microscopy after 24 h of FK228 (5 nM) treatment. Cells were stained for ANXA1 (by monoclonal or polyclonal antibody, green), and nuclei were counterstained with DAPI (blue). Results are representative of two independent experiments. (b) FK228-induced ANXA1 cleavage and caspase-3 cleavage in Kasumi-1 cells. Whole-cell lysates were prepared from Kasumi-1 cells cultured with and without FK228 (5 nM) or FK228/ Z-DEVD-fmk (50 μM) for 24 h and subjected to immunoblot analysis for ANXA1 and caspase-3 expression. The anti-polyclonal and monoclonal ANXA1, anti-procaspase-3, and anti-cleaved caspase-3-antibodies were used. β-actin showed equal loading of samples. The data shown are representative of three independent experiments. (c) Neutralization of ANXA1 inhibits FK228-induced apoptosis in Kasumi-1 cells. Percentage of Kasumi-1 cells showing FK228-induced annexin V-positivity pretreated with or without ANXA1 neutralizing antibody. FK228 was added to the cells 24 h following pretreatment with the ANXA1 neutralizing antibody, after which cells were washed and the medium changed. Cells were then incubated for 48 h, and annexin V positivity was analyzed by flow cytometry. Graphs show the mean±S.D. of results from three independent experiments. Statistically significant differences (*P<0.05) were determined by a paired Student's t-test
To ascertain the contribution of ANXA1 upregulation to FK228-induced apoptosis, we first examined the effect of the ANXA1 neutralizing antibody on the cytotoxic effects of FK228 on Kasumi-1 cells. The neutralization of ANXA1 completely blocked FK228-induced apoptosis in Kasumi-1 cells (Figure 4c). In an alternative approach, we analyzed FK228-induced apoptosis in ANXA1 knockdown Kasumi-1 cells. This showed that ANXA1 mRNA expression in ANXA1 siRNA-transfected cells was reduced by 80% at 48 h as compared with its expression in non-specific (NS) siRNA-transfected cells (Figure 5a, i). This translated into the complete disappearance of the cleaved isoform of ANXA1 and a partial decrease in the expression of the full-length protein (Figure 5a, ii). Notably, ANXA1 siRNA-transfected cells were significantly less susceptible to FK228-induced apoptosis than were NS-siRNA-transfected cells (Figure 5b, i). However, ANXA1 silencing only partially blocked the growth-inhibitory effects of FK228 (Figure 5b, ii), suggesting that while ANXA1 plays a role in apoptosis induction, ANXA1-independent mechanisms are responsible for the anti-proliferative effects of FK228.

(a) ANXA1 knockdown in ANXA1 siRNA-transfected Kasumi-1 cells. Kasumi-1 cells were transfected with NS-siRNA or ANXA1 siRNA, as described in Materials and Methods. (i) The abundance of transcripts of ANXA1 mRNA relative to that of β2-microglobulin in control (untransfected) and NS-siRNA- or ANXA1 siRNA-transfected Kasumi-1 cells is shown. Graphs show the mean±S.D. of representative data from three independent experiments. A statistically significant difference (**P<0.01) was determined by a paired Student's t-test. (ii) ANXA1 protein expression in NS-siRNA- or ANXA1 siRNA-transfected Kasumi-1 cells was analyzed in Western blots using anti-rabbit polyclonal-ANXA1 antibody. The data are representative of two experiments that produced comparable results. (b) Effects of ANXA1 silencing by siRNA on apoptosis and viability of Kasumi-1 cells. Percentage of annexin V-positive cells (and viable cell counts (ii) for NS-siRNA- or ANXA1 siRNA-transfected Kasumi-1 cells treated with FK228. FK228 was added for 24 h, after which cells were washed, the medium changed, and cells incubated for 48 h. Annexin V positivity was measured by flow cytometry, and viable cell counts were determined using the Trypan blue exclusion method. Graphs show the mean±S.D. of results from three independent experiments. Statistically significant differences (*P<0.05) were determined by a paired Student's t-test
ANXA1 mediates engulfment of apoptotic cells by macrophages
Since ANXA1 is an endogenous ligand that naturally mediates the engulfment of apoptotic cells,21 we investigated the functional effects of ANXA1 on the phagocytosis of leukemia cells. We used a co-culture system consisting of human THP-1-derived macrophages and Kasumi-1 cells and performed a cell adherence assay to evaluate the engulfment of Kasumi-1 cells by macrophages in co-culture. As shown in Figure 6a(i), exposure to FK228 for 24 h dramatically increased the attachment of Kasumi-1 cells to macrophages (untreated versus FK228-treated cells: 57±9 cells versus 196±33 cells/microscopic fields (0.08 mm2/field), n=5; P=0.01). The engulfment of Kasumi-1 cells by macrophages was confirmed by fluorescence-labeling experiments, in which macrophages were labeled with PE-conjugated CD14 (Figure 6a, ii).


Involvement of ANXA1 in engulfment of Kasumi-1 cells by macrophages. Clustering of FK228 (5 nM)-treated Kasumi-1 cells on macrophage-differentiated THP-1. Control and FK228-treated Kasumi-1 cells were added to the macrophage- differentiated THP-1 for 3 h. Kasumi-1 cell clustering was visualized after wash-out of non-ingested cells. (i) Representative photomicrograph of Kasumi-1 cell clustering on the macrophage-differentiated THP-1 monolayer (× 40). (ii) The engulfment by macrophages was visualized by immunofluoresence detection of macrophages with PE-conjugated anti-CD14 mouse monoclonal antibody (× 400). (b) Kasumi-1 cells transfected with ANXA1 siRNA or NS siRNA and treated with FK228 were co-cultured with macrophage-differentiated THP-1 for 3 h. Representative photomicrograph of Kasumi-1 cells clustering on the macrophage-differentiated THP-1 monolayer: (i) × 40 and (ii) × 400. (c) Kasumi-1 cells pretreated with ANXA1 neutralizing antibody followed by FK228 were co-cultured with macrophage-differentiated THP-1 for 3 h. Representative photomicrograph of Kasumi-1 cells clustering on the macrophage-differentiated THP-1 monolayer: (i) × 40 and (ii) × 400
In contrast, FK228-induced cell attachment was markedly abrogated in the ANXA1 siRNA-transfected Kasumi-1 cells (60.5±10.5% inhibition; n=5; P<0.001; Figure 6b). Likewise, pretreatment with FK228 and ANXA1 neutralizing antibody for 24 h before the co-culture experiments resulted in the significant repression of FK228-stimulated engulfment of leukemia cells by macrophages (54.1±3.0% inhibition; n=5; P=0.02; Figure 6c). However, this effect was not enhanced when ANXA1 neutralizing antibody was added to the medium at the time of co-culture, suggesting that it is the membrane-bound and not soluble form of ANXA1 that contributes to the engulfment of leukemia cells by macrophages (not shown).
Discussion
In this study, we investigated a novel mechanism of action of HDACIs in leukemia cells harboring the AML1-ETO-fusion protein. In particular, we observed a profound growth inhibition and apoptosis of AML1-ETO-positive Kasumi-1 and SKNO-1 cells in response to FK228 and SAHA. We further demonstrated that this response was associated with upregulation of the pro-apoptotic gene ANXA110, 11, 12, 22 through histone acetylation in its promoter region. In contrast, HDAC inhibition failed to induce ANXA1 expression in the APL cell line NB4 or in the other AML lines U937 and OCI-AML2. These AML1-ETO-negative cells also showed a decreased sensitivity to the growth-inhibitory effects of HDACIs, suggesting that ANXA1 contributes to the pro-apoptotic effects of these agents.
The specific modulation of the ANXA1 transcription in AML1-ETO-positive cells suggests a molecular link between this chimeric fusion protein and the regulation of ANXA1 promoter activity. As it is currently understood, AML1-ETO recruits the HDAC complex to AML1-dependent promoters,1, 4, 23 which results in the transcriptional modulation of AML1-regulated genes, including the upregulation of TIS11b,24 c-myc, cyclin D125 genes, and downregulation of the genes that encode the transcription factor C/EBPα,19, 20 the tumor suppressor p14ARF,26 the neurofibromatosis-1 protein,27 and interleukin 3.28 Recently, the proteasomal degradation of AML1-ETO by FK228 was reported to result from the interrupted association of AML1-ETO with heat shock protein 90 (HSP90).29 Adding to this understanding, our studies identified C/EBPα binding sites within the ANXA1 promoter region that contain no direct AML1-binding site. Our findings that FK228 upregulated the C/EBPα gene and promoted C/EBPα binding on the ANXA1 promoter indicate the possibility that C/EBPα-dependent ANXA1 transcriptional activation is one of the molecular mechanisms dictating the specificity of histone acetylation and transcriptional activation of the target genes in AML1-ETO-expressing AML cells. However, our findings of apoptosis induction by high doses of FK228 or SAHA independent of AML1-ETO-expression, and the observation that ANXA1 silencing in Kasumi-1 cells only partially blocked growth-inhibitory effects of FK228, suggest that while ANXA1 plays a role in apoptosis induction in AML1-ETO-expressing Kasumi-1 cells, ANXA1-independent mechanisms may contribute to pro-apoptotic properties of HDACIs irrespective of AML1-ETO expression. Recent reports have attributed the anti-leukemia activity of the HDACIs SAHA, valproic acid, and the benzamide derivative MS275 to the direct activation of the promoter of the tumor-selective death ligand TRAIL through the cAMP response element binding protein (CBP) and the recruitment of SP1 and SP3 in AML cells.7 These effects were seen in AML cells irrespective of their AML-ETO expression status. Since, similar to MS275, FK228 also potently inhibits HDAC1 and HDAC2,30 the induction of TRAIL may also contribute to the FK228-induced apoptosis in Kasumi-1 cells. The recruitment of CBP by the HDACI in turn induces promoter histone acetylation and the target gene transcription.4 Although the ANXA1 promoter does not contain an SP1 site, the role of CBP recruitment in the induction of ANXA1 and identification of the acetylation sites involved await further characterization.
We next focused on the pro-apoptotic function of ANXA1 in AML1-ETO-expressing AML cells. In this regard, FK228 was found to increase the protein levels of both the full-length and 33 kDa N-terminal cleavage products of ANXA1, which led to the localization of ANXA1 on the plasma membrane. Conversely, the inhibition of ANXA1 function (by ANXA1 neutralizing antibody, which blocked ANXA1 localization on the plasma membrane) or expression (by siRNA) significantly reduced FK228-induced apoptosis, indicating that ANXA1 is involved in apoptosis induction in response to FK228. In addition, our results suggest a specific role for the cleaved ANXA1 protein in accelerating the cell death process. Previous reports indicated that cells transfected with cleaved ANXA1 protein showed higher constitutive and inducible caspase-3 activity,12 suggesting that activation of ANXA1 contributes to the initiation of the caspase cascade and apoptosis. We demonstrated that, in turn, caspase-3 inhibition partially blocks FK228-induced apoptosis and ANXA1 cleavage without upregulating the ANXA1 gene. This finding is in agreement with that of Debret et al.,11 who demonstrated that ANXA1 cleavage was inhibited in U937 cells by caspase inhibitors. These findings suggest that although ANXA1 contributes to caspase-3 activation, cleaved caspase-3 in turn promotes ANXA1 cleavage and apoptosis in leukemia cells.
Our cDNA array data demonstrated that FK228 upregulates 123 genes, plus it cooperates with other proteins in the upregulation of the genes that encode the pro-apoptotic proteins TNFα (2.1-fold) and profilin (four-fold) that have been reported to associate with ANXA1.12, 31 It has been further shown that FK228 activates TNFα gene transcription through the hyperacetylation of histone H3 and H4 in its promoter region, which is associated with the activation of caspases-8 and -10 and hence the cleavage of caspases-3 and -7.16 Furthermore, the treatment of U937 cells with TNFα has been shown to increase ANXA1 mRNA and protein expression;12 vice versa, U937 cells overexpressing ANXA1 have been reported to be more susceptible to TNFα-induced apoptosis.32 These observations suggest that ANXA1 and TNFα, whose gene promoters are the targets of FK228-induced histone acetylation, cooperatively promote apoptosis induction. The transcriptional activation of several other proapoptotic genes, including Fas, Bax, and TRAIL, has also been implicated in HDAC-induced apoptosis.7, 33 Some of these FK228-induced apoptotic events may occur in association with the activation of caspase-3 that contributes to increased ANXA1 expression and cleavage.
In addition to the role of ANXA1 in the apoptosis induced by FK228, our studies identified a unique role for ANXA1 in the interactions between leukemia cells and macrophages, one of the critical attributes of the bone marrow microenvironment. Specifically, the ANXA1 neutralizing antibody and siRNA/ANXA1 effectively blocked the engulfment of FK228-treated Kasumi-1 cells by macrophages, suggesting that membrane-localized ANXA1 plays a critical role in phagocytosis. This is potentially explained by the fact that the apoptotic cells induced by antitumor therapy must be efficiently recognized and internalized by macrophages. In keeping with this, Arur et al.21 demonstrated that ANXA1 was recruited from the cytosol to the membrane in Jurkat T cells undergoing apoptosis and that this recruitment was specific to ANXA1 among the annexins expressed. These authors concluded from this that the export of ANXA1 was a critical signal for the engulfment of the apoptotic cells.
While these observations document the role for ANXA1 as a caspase-dependent endogenous ligand that mediates the engulfment of apoptotic cells and hence serves as a bridging molecule between apoptotic cells and macrophages, ours is the first observation that HDACIs engage in this mechanism. In line with these findings, the ANXA1-derived peptide Ac2-26, which corresponds to the first amino acids of the ANXA1 N terminus, was recently shown to stimulate the phagocytosis of apoptotic polymorphonuclear leukocytes by human monocyte-derived macrophages.34 Our cDNA array data showed that the anti-inflammatory cytokine TGF-β, which functions in conjunction with ANXA1 in phagocytosis,17 was also transcriptionally induced by FK228, suggesting the close interplay between ANXA1 and TGF-β in the phagocytosis of apoptotic Kasumi-1 cells.
In summary, our findings suggest that FK228 and SAHA, a highly potent antitumor agent currently undergoing phase I/II clinical trials in patients with hematologic malignancies,9, 35 induce the transcriptional activation of ANXA1 through the histone acetylation of its promoter. Our data further suggest that the induction of ANXA1 is specific to leukemia cells expressing the AML1-ETO chimeric fusion protein. This results in the upregulation and externalization of the intracellular ANXA1 protein that induces apoptosis and, more importantly, provides an ‘eat-me’ signal that leads to the phagocytic clearance of the leukemia cells by macrophages. Since the rapid recognition and engulfment of apoptotic cells by neighboring macrophages and phagocytosis is an important component of anticancer therapy, we conclude that FK228 has promise as an effective antitumor agent in AML1-ETO-positive leukemias, functioning via dual mechanisms-the induction of apoptosis and the control of inflammatory responses.
Materials and Methods
Reagents and cell cultures
We used the following two AML1-ETO-positive AML cell lines: Kasumi-1, which was provided by Dr. H. Asou,36 and SKNO-1, which was provided by Dr. T. Nakagawa.37 The following four AML1-ETO-negative cell lines were also obtained. The NB4 cell line was provided by Dr. M. Lanotte,38 and the OCI-AML2 cell line was provided by Dr. M. Minden (Ontario Cancer Institute, Toronto, Canada). U937 cells were purchased from the American Type Culture Collection (Rockville, MD, USA), and THP-1 cells were purchased from Riken Bioresource Center (Tsukuba, Japan). Kasumi-1, NB4, OCI-AML2, U937, and THP-1 cells were maintained in RPMI-1640 medium containing 10% fetal calf serum, 1% L-glutamine, and penicillin–streptomycin; 2 ng/ml GM-CSF was added to SKNO-1 cell cultures. FK228 (Fujisawa Pharmaceutical Co. Ltd., Osaka, Japan), which was dissolved in dimethylsulfoxide to a concentration of 10 mM and stored at −20°C, was diluted in culture medium before the in vitro exposure of cells.
Cells were treated with FK228, the caspase-3 inhibitor Z-DEVD-fmk (Calbiochem, La Jolla, CA, USA), and/or SAHA (provided by Drs. P. Marks and V. Richon, Merck & Co. Inc., Whitehouse Station, NJ, USA) at the indicated concentrations. Medium was changed after 16 h of FK228 exposure.
Cell viability assay
At 24–96 h after drug exposure, viable cells were identified using the Trypan blue dye exclusion method and counted in a hematocytometer.
Flow cytometric analysis
The cell cycle distribution was determined by flow cytometry of propidium iodide (PI)-stained nuclei. Briefly, cells were washed twice with phosphate-buffered saline (PBS), fixed in ice-cold ethanol (70% v/v in water), and stained with the PI solution (25 μg/ml PI, 180 U/ml RNase, 0.15% Triton X-100, and 30 mg/ml polyethylene glycol in 4 mM citrate buffer, pH 7.8; all from Sigma Chemical Co., St. Louis, MO, USA). The DNA content was measured using a FACScan flow cytometer (Becton Dickinson Immunocytometry Systems, San Jose, CA, USA) and analyzed by the ModFit program (Verity Software House, Topsham, ME, USA). Cells with a hypodiploid DNA content (<2n) were considered apoptotic.
For the PI/annexin V-binding studies done to analyze apoptosis, fresh cells were washed twice with binding buffer (10 mM HEPES, 140 mM NaCl, and 5 mM CaCl2, pH 7.4; all from Sigma Chemical Co.) and stained with FITC-conjugated annexin V (Roche Diagnostic Co., Indianapolis, IN, USA) for 15 min at room temperature. Annexin V fluorescence was determined using a Becton Dickinson FACScan flow cytometer, and the integrity of the cell membranes was simultaneously assessed by the PI exclusion method.
Oligonucleotide array-based expression profiling
Hybridization was performed using human genome U95Av2 probe arrays (Affymetrix, Santa Clara, CA, USA) containing probe sets with previously characterized genes. The target was labeled and hybridized to the probe arrays, and the arrays washed, stained, and scanned, as described previously. Briefly, total RNAs were prepared after 24 h of treatment with FK228 (5 nM) using the RNeasy Total RNA isolation kit (Qiagen, Valencia, CA, USA), and double-stained cDNA was synthesized from total RNA. An in vitro transcription reaction was carried out to produce biotin-labeled cRNA from the cDNA, and the cRNA was fragmented and hybridized to the oligonucleotide probes on the probe array during 16 h of incubation at 45°C. Immediately after hybridization, the hybridized probe array underwent an automated washing and staining on the fluids station, and the Genechip Microarray Suite 4.0 Software (Affymetrix) was used to measure the intensity of the expression of each target gene by calculating the average differences (perfect match intensity minus mismatch intensity) of the 14–20 probe pairs used for the particular gene.
DNA-Chip Analyzer Software was used to analyze the quantified images and the expression change for each target gene was estimated with the reduced Li Wong PM-MM difference model.39 The statistical results were imported into the Result Viewer 2.0 software.
Western blot and antibodies
An equal amount of cell lysate was separated by 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis, followed by immunoblotting on Hybond-P membranes (Amersham Pharmacia Biotech, Buckinghamshire, UK). Proteins were visualized using the enhanced chemiluminescence detection system (Amersham Pharmacia Biotech) after incubation overnight at 4°C with the following antibodies: anti-rabbit polyclonal ANXA1 antibody,15 anti-mouse monoclonal ANXA1 antibody (Santa Cruz Biotechnology, Santa Cruz, CA, USA), and anti-procaspase-3 and anti-cleaved caspase-3 antibodies (Cell Signaling Technology, Beverly, MA, USA). Anti-β-actin (Abcam, Cambridge, MA, USA) blots were run in parallel as loading controls.
Quantitative real-time PCR
RNA was isolated using TRIZOL (Invitrogen Co., Carlsbad, CA, USA). One microgram of total RNA template was used per 10 μl of reverse transcriptase reaction by AMV reverse transcriptase (Roche Diagnostic Co.) using hexanucleotide random primers at 42°C for 1 h following the manufacturer's instructions. For quantitative real-time RT-PCR, duplicate 1 μl samples of each cDNA were amplified as follows: 50°C for 2 min, 95°C for 10 min, 40 or 50 cycles at 95°C for 15 s, and 60°C for 60 s. The relative amount of gene expression was calculated using the expression of β2-microglobin as an internal standard. Primers (FP, forward; RP, reverse) and probe sequences of ANXA1, C/EBPα, and β2-microglobin (β2-m) were as follows: ANXA1 (GenBankδ accession number BC035993; nucleotides 375–450), FP: 5′-CAGGTCACCTTGAGGAGGTTGT-3′; RP: 5′-CAGCACGAAGTTCATCAGCATC-3′; probe: 5′-TTGGCTCTGCTAAAAACTCCAGCGCA-3′; C/EBPα (GenBank™ accession number NM_004364; nucleotides 161–230) FP:5′- GCATCTGCGAGCACGAGAC-3′; RP:5′- AACTCGTCGTTGAAGGCGG-3′; probe:5′- TCCATCGACATCAGCGCCTACATCG-3′; β2-m FP: 5′-AGCTGTGCTCGCGCTACTCT-3′; RP: 5′-TTGACTTTCCATTCTCTGCTGG-3′; probe: 5′-TCTTTCTGGCCTGGAGGGCATCC-3′. The abundance of each transcript of interest relative to the abundance of β2-microglobulin was calculated as follows: relative expression (RE)=100 × 2exp(−ΔCT), where ΔCT is the mean CT of the transcript of interest minus the mean CT of the transcript for β2-microglobulin. To obtain the mean CT values, we performed all reactions in duplicate.
Chromatin immunoprecipitation assay
The chromatin immunoprecipitation (ChIP) assay was performed following the manufacturer's instructions (Upstate Biotechnology, Lake Placid, NY, USA). The cells were fixed with formaldehyde (final concentration, 1%), lysed, sonicated, and then immunoprecipitated by overnight incubation at 4°C with anti-acetylated H3-lys9, anti-dimethylated H3-Lys9 (both antibodies from Upstate Biotechnology), and anti-C/EBPα (Santa Cruz Biotechnology). PBS or rabbit IgG was added as a negative control. A small portion of chromatin was set aside for use as the ‘input’ fraction by processing for immunoprecipitated chromatin. After immunoprecipitation, salmon sperm DNA beads (Upstate Biotechnology) were added and incubated for 1 h at 4°C. The chromatin-antibody/beads complexes were washed sequentially with low salt, high salt, and LiCl immune complex wash buffers and then with TE buffer. The immunoprecipitated and input chromatin was treated with RNase and proteinase K and maintained at 65°C for 6 h to reverse the formaldehyde crosslinks, after which the DNA fragments were purified (QIAquick PCR purification kit; Qiagen, Tokyo, Japan). Total DNA (10 × diluted input fraction) and immunoprecipitated DNA samples were applied to real-time PCR. For quantitative real-time PCR, primers and probes were designed by Primer Express software packaged with the Applied Biosystems Model 7700 sequence detector (PE Applied Biosystems, Foster City, CA, USA). Probes and primers were selected to include the proximal promoter where a large number of the described transcriptional response elements reside. The probes were labeled at the 5′ end with FAM and at the 3′ end with TAMRA, which served as a quencher. The amplification conditions were as follows: 50°C for 2 min, 95°C for 10 min, 50 cycles at 95°C for 15 s, and 60°C for 60 s. Annexin A1 promoter elements (GenBank™ accession number U25414; nucleotides 873–953), FP: 5′-TCACTTTGTTTTTGGACATAGCTGA-3′; RP: 5′-CCACACCTAGCAACCAGAAGTTAG-3′; probe: 5′-CCATGTACTTCAAACAGAAGGCAGCCAATT-3′. The abundance of each transcript of interest relative to that of ‘input’ CT values was calculated as follows: , where ΔCT is the difference in CT between the transcript of interest and total DNA (10 × diluted ‘input’ fraction), which represents a relative change of the targeted modification in the ANXA1 gene promoter region compared with the input.40 The CT data from duplicate PCRs, in which the same DNA was used were averaged before the RE was calculated.
The Gene regulation program (www.gene-regulation.com) was used to predict transcription factor binding sites.
Small interfering RNA transfection
ANXA1 gene expression in leukemia cells was silenced by the siRNA technique. The sense strand of the siRNA silencing (ANXA1-siRNA) was ACUCCAGCGCAAUUUGAUGTT (BC035993; nucleotides 412–432). Mock transfection was done with buffer alone. A nonspecific control pool containing four pooled nonspecific siRNA duplexes was also used as a negative control (referred to as NS-siRNA; Takara Bio Inc., Shiga, Japan). Transfection of Kasumi-1 cells was facilitated by the Trans IT-TKO Tranfection Reagent (Mirus Corporation, Madison, WI, USA), following the manufacturer's instructions, with modification. Briefly, 24 μl of the TKO reagent was first incubated with 100 μl of OPTIMEM-I medium (Invitrogen Co.) at room temperature for 15 min before siRNA was added. After another 15 min at room temperature, 500 μl of RPMI-1640 (with 20% calf serum) containing 0.8 × 106 cells was added. The final siRNA concentration was 125 nM, and the incubation was allowed to continue for 4 h at 37°C in 5% CO2, after which 3.5 ml of RPMI-1640 (with 20% calf serum) was added, and the incubation continued at 37°C for 24 s. FK228 was added to a final concentration, as indicated, and the cells were harvested 72 h later for apoptosis and cell cycle analysis. The gene silencing was verified by TaqMan RT-PCR and Western blot performed at 48 h after transfection.
Preparation of cells for phagocytosis studies
THP-1 cells (0.5 × 106 cells per 35-mm dish) were first differentiated by stimulation with PMA (160 nM) for 72 h to obtain a macrophage-like phenotype that closely resembled that of human monocyte-derived macrophages. Kasumi-1 cells were cultured at a density of 0.2 × 106 cells/ml in the presence or absence of FK228 (5 nM) for 24 h, after which the medium was changed. PMA-treated THP-1 cells were washed with PBS, and Kasumi-1 cells (either treated or untreated with FK228) added. Cells were then incubated for 3 h at 37°C. Non-ingested cells were removed by three washes with cold PBS. The engulfment by macrophages was visualized by the immunofluorescence detection of macrophages with PE-conjugated anti-CD14 mouse monoclonal antibody.
For each experiment, the number of Kasumi-1 cells that adhered to macrophages was determined in at least five fields (× 200), and an average was calculated for duplicate wells.
For the ANXA1-neutralizing studies, macrophage-differentiated THP-1 and Kasumi-1 cells were treated for 24 h with the neutralizing anti-rabbit polyclonal ANXA1 antibody (1 : 100) that binds to full-length and N-terminal–truncated ANXA1 with and without FK228 before co-incubation, as described above.
Confocal microscopy
In preparation for confocal microscopy, cells were washed with PBS, fixed with 4% paraformaldehyde in PBS for 10 min at room temperature, permeabilized with 90% methanol in TBST buffer (10 mM Tris, 15 mM NaCl, and 0.1% Triton X-100) for 15 min, and rinsed with PBS for 5 min. Next, cells were blocked with 5% goat serum in PBS for 30 min and incubated with anti-mouse monoclonal ANXA1 antibody (1 : 100) or anti-rabbit polyclonal ANXA1 antibody (1 : 100) overnight at 4°C. Excess antibody was removed by washing with PBS. Then cells were incubated with secondary FITC-labeled anti-mouse IgG (1 : 250, H+L; Calgat, Burlingame, CA, USA) for 30 min at 37°C. Cells were washed with PBS, mounted on slides, and analyzed under a Zeiss LSM 510 laser confocal microscope (Carl Zeiss, Thornwood, NY, USA).
Statistical analysis
The results are expressed as the mean±SD. Levels of significance were evaluated by a two-tailed, paired, Student's t-test, and P<0.05 was considered statistically significant.
References
Wang J, Hoshino T, Redner RL, Kajigaya S, Liu JM . ETO, fusion partner in t(8;21) acute myeloid leukemia, represses transcription by interaction with the human N-CoRmSin3HDAC1 complex. Proc Natl Acad Sci USA 1998; 95: 10860–10865.
Strahl B, Allis CD . The language of covalent histone modifications. Nature 2000; 403: 41–45.
Khochbin S, Verdel A, Lemercier C, Seigneurin-Berny D . Functional significance of histone deacetylase diversity. Curr Opin Genet Dev 2001; 11: 162–166.
Redner RL, Wang J, Liu JM . Chromatin remodeling and leukemia: new therapeutic paradigms. Blood 1999; 94: 417–428.
Bhalla KN . Epigenetic and chromatin modifiers as targeted therapy of hematologic malignancies. J Clin Oncol 2005; 23: 3971–3993.
Kosugi H, Ito M, Yamamoto Y, Towatari M, Ito M, Ueda R et al. In vivo effects of a histone deacetylase inhibitor, FK228, on human acute promyelocytic leukemia in NOD / Shi-scid/scid mice. Jpn J Cancer Res 2001; 92: 529–536.
Nebbioso A, Clarke N, Voltz E, Germain E, Ambrosino C, Bontempo P et al. Tumor-selective action of HDAC inhibitors involves TRAIL induction in acute myeloid leukemia cells. Nat Med 2005; 11: 77–84.
Piekarz RL, Robey RW, Zhan Z, Kayastha G, Sayah A, Abdeldaim AH et al. T-cell lymphoma as a model for the use of histone deacetylase inhibitors in cancer therapy: impact of depsipeptide on molecular markers, therapeutic targets, and mechanisms of resistance. Blood 2004; 103: 4636–4643.
O'Connor OA, Heaney ML, Schwartz L, Richardson S, Willim R, MacGregor-Cortelli B et al. Clinical experience with intravenous and oral formulations of the novel histone deacetylase inhibitor suberoylanilide hydroxamic acid in patients with advanced hematologic malignancies. J Clin Oncol 2006; 24: 166–173.
Garcia Pedrero JM, Fernandez MP, Morgan RO, Herrero Zapatero A, Gonzalez MV, Suarez Nieto C et al. Annexin A1 down-regulation in head and neck cancer is associated with epithelial differentiation status. Am J Pathol 2004; 164: 73–79.
Debret R, El Btaouri H, Duca L, Rahman I, Radke S, Haye B et al. Annexin A1 processing is associated with caspase-dependent apoptosis in BZR cells. FEBS Lett 2003; 546: 195–202.
Solito E, de Coupade C, Canaider S, Goulding NJ, Perretti M . Transfection of annexin 1 in monocytic cells produces a high degree of spontaneous and stimulated apoptosis associated with caspase-3 activation. Br J Pharmacol 2001; 133: 217–228.
Raynal P, Pollard HB . Annexins: the problem of assessing the biological role for a gene family of multifunctional calcium- and phospholipid-binding proteins. Biochim Biophys Acta 1994; 1197: 63–93.
Solito E, Kamal A, Russo-Marie F, Buckingham JC, Marullo S, Perretti M . A novel calcium-dependent proapoptotic effect of annexin 1 on human neutrophils. FASEB J 2003; 17: 1544–1546.
Borisenko GG, Matsura T, Liu SX, Tyurin VA, Jianfei J, Serinkan FB et al. Macrophage recognition of externalized phosphatidylserine and phagocytosis of apoptotic Jurkat cells – existence of a threshold. Arch Biochem Biophys 2003; 413: 41–52.
Sutheesophon K, Nishimura N, Kobayashi Y, Furukawa Y, Kawano M, Itoh K et al. Involvement of the tumor necrosis factor (TNF)/TNF receptor system in leukemic cell apoptosis induced by histone deacetylase inhibitor depsipeptide (FK228). J Cell Physiol 2005; 203: 387–397.
Fadok VA, Bratton DL, Konowal A, Freed PW, Westcott JY, Henson PM . Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGF-β, PGE2, and PAF. J Clin Invest 1998; 101: 890–898.
Lutterbach B, Westendorf JJ, Linggi B, Patten A, Moniwa M, Davie JR et al. ETO, a target of t(8;21) in acute leukemia, interacts with the N-Cor and mSin3 corepressors. Mol Cell Biol 1998; 18: 7176–7184.
Westendorf JJ, Yamamoto CM, Lenny N, Downing JR, Selsted ME, Hiebert SW . The t(8;21) fusion product, AML1-ETO, associates with C/EBPα, inhibits C/EBPa-dependent transcription, and blocks granulocytic differentiation. Mol Cell Biol 1998; 18: 322–333.
Pabst T, Mueller BU, Harakawa N, Schoch C, Haferlach T, Behre G et al. AML1-ETO downregulates the granulocytic differentiation factor C/EBPalpha in t(8;21) myeloid leukemia. Nat Med 2001; 7: 444–451.
Arur S, Uche UE, Rezaul K, Fong M, Scranton V, Cowan AE et al. Annexin I is an endogenous ligand that mediates apoptotic cell engulfment. Dev Cell 2003; 4: 587–598.
Perretti M, Solito E . Annexin 1 and neutrophil apoptosis. Biochem Soc Trans 2004; 32: 507–510.
Gelmett V, Zhang J, Fanelli M, Minucci S, Pelicci PG, Lazar MA . Aberrant recruitment of the nuclear receptor corepressor−histone deacetylase complex by the acute myeloid leukemia fusion partner ETO. Mol Cell Biol 1998; 18: 7185–7191.
Shimada H, Ichikawa H, Nakamura S, Katsu R, Iwasa M, Kitabayashi I et al. Analysis of genes under the downstream control of the t(8;21) fusion protein AML1-MTG8: overexpression of the TIS11b (ERF-1, cMG1) gene induces myeloid cell proliferation in response to G-CSF. Blood 2000; 96: 655–663.
Muller-Tidow C, Steffen B, Cauvet T, Tickenbrock L, Ji P, Diederichs S et al. Translocation products in acute myeloid leukemia activate the Wnt signaling pathway in hematopoietic cells. Mol Cell Biol 2004; 24: 2890–2904.
Linggi B, Muller-Tidow C, van de Locht L, Hu M, Nip J, Serve H et al. The t(8;21) fusion protein, AML1 ETO, specifically represses the transcription of the p14(ARF) tumor suppressor in acute myeloid leukemia. Nat Med 2002; 8: 743–750.
Yang G, Khalaf W, van de Locht L, Jansen JH, Gao M, Thompson MA et al. Transcriptional repression of the Neurofibromatosis-1 tumor suppressor by the t(8;21) fusion protein. Mol Cell Biol 2005; 25: 5869–5879.
Uchida H, Zhang J, Nimer SD . AML1A and AML1B can transactivate the human IL-3 promoter. J Immunol 1997; 158: 2251–2258.
Yang G, Thompson MA, Brandt SJ, Hiebert SW . Histone deacetylase inhibitors induce the degradation of the t(8;21) fusion oncoprotein. Oncogene 2007; 26: 91–101.
Furumai R, Matsuyama A, Kobashi N, Lee KH, Nishiyama M, Nakajima H et al. FK228 (depsipeptide) as a natural prodrug that inhibits class I histone deacetylases. Cancer Res 2002; 62: 4916–4921.
Alvarez-Martinez MT, Porte F, Liautard JP, Sri Widada J . Effects of profilin-annexin I association on some properties of both profilin and annexin I: modification of the inhibitory activity of profilin on actin polymerization and inhibition of the self-association of annexin I and its interactions with liposomes. Biochim Biophys Acta 1997; 1339: 331–340.
Canaider S, Solito E, DeCoupade C, Flower RJ, Russo-Marie F, Goulding NJ et al. Increased apoptosis in U937 cells over-expressing lipocortin 1 (annexin I). Life Sci 2000; 66: L265–L270.
Kwon SH, Ahn SH, Kim YK, Bae G-U, Yoon JW, Hong S et al. Apicidin, a histone deacetylase inhibitor, induces apoptosis and Fas/Fas ligand expression in human acute promyelocytic leukemia cells. J Biol Chem 2002; 277: 2073–2080.
Reville K, Crean JK, Vivers S, Dransfield I, Godson C . Lipoxin A4 redistributes myosin IIA and Cdc42 in macrophages: implications for phagocytosis of apoptotic leukocytes. J Immunol 2006; 176: 1878–1888.
Byrd JC, Marcucci G, Parthun MR, Xiao JJ, Klisovic RB, Moran M et al. A phase 1 and pharmacodynamic study of depsipeptide (FK228) in chronic lymphocytic leukemia and acute myeloid leukemia. Blood 2005; 105: 959–967.
Asou H, Tashiro S, Hamamoto K, Otsuji A, Kita K, Kamada N . Establishment of a human acute myeloid leukemia cell line (Kasumi-1) with 8;21 chromosome translocation. Blood 1991; 77: 2031–2036.
Matozaki S, Nakagawa T, Kawaguchi R, Aozaki R, Tsutsumi M, Murayama T et al. Establishment of a myeloid leukaemic cell line (SKNO-1) from a patient with t(8;21) who acquired monosomy 17 during disease progression. Br J Haematol 1995; 89: 805–811.
Lanotte M, Martin-Thouvenin V, Najman S, Balerini P, Valensi F, Berger R . NB4, a maturation inducible cell line with t(15;17) marker isolated from a human acute promyelocytic leukemia (M3). Blood 1991; 77: 1080–1086.
Li C, Wong WH . Model-based analysis of oligonucleotide arrays: expression index computation and outlier detection. Proc Natl Acad Sci USA 2001; 98: 31–36.
Baguet A, Sun X, Arroll T, Krumm A, Bix M . Intergenic transcription is not required in Th2 cells to maintain histone acetylation and transcriptional permissiveness at the Il4-Il13 locus. J Immunol 2005; 175: 8146–8153.
Acknowledgements
We thank Betty Notzon for manuscript review, Nobuko Tanaka, Hiroaki Miyajima, and Akemi Koyanagi for technical assistance, and Rosemarie Lauzon for administrative assistance in the preparation of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
Edited by R De Maria
Rights and permissions
About this article
Cite this article
Tabe, Y., Jin, L., Contractor, R. et al. Novel role of HDAC inhibitors in AML1/ETO AML cells: activation of apoptosis and phagocytosis through induction of annexin A1. Cell Death Differ 14, 1443–1456 (2007). https://doi.org/10.1038/sj.cdd.4402139
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.cdd.4402139
Keywords
This article is cited by
-
Multiple Mild Stimulations Reduce Membrane Distribution of CX3CR1 Promoted by Annexin a1 in Microglia to Attenuate Excessive Dendritic Spine Pruning and Cognitive Deficits Caused by a Transient Ischemic Attack in Mice
Neuroscience Bulletin (2022)
-
AML1/ETO and its function as a regulator of gene transcription via epigenetic mechanisms
Oncogene (2021)
-
The ubiquitin ligase RNF5 determines acute myeloid leukemia growth and susceptibility to histone deacetylase inhibitors
Nature Communications (2021)
-
Immune system deregulation in hypertensive patients chronically RAS suppressed developing albuminuria
Scientific Reports (2017)
-
Strong Correlation between the Expression Levels of HDAC4 and SIRT6 in Hematological Malignancies of the Adults
Pathology & Oncology Research (2017)
