
Abstract
Reactive oxygen species (ROS) have been closely associated with both apoptotic and non-apoptotic/necrotic cell death. Our previous study has illustrated that c-Jun-N-terminal kinase 1 (JNK1) is the main executor in hydrogen peroxide (H2O2)-induced nonapoptotic cell death. The main objective of this study is to further elucidate the molecular mechanisms downstream of JNK1 in H2O2-induced cell death. In this study, poly(ADP-ribose) polymerase-1 (PARP-1), a key DNA repair protein, was readily activated by H2O2 and inhibition of PARP-1 activation by either a pharmacological or genetic approach offered significant protection against H2O2-induced cell death. More importantly, H2O2-mediated PARP-1 activation is subject to regulation by JNK1. Suppression of JNK1 activation by a chemical inhibitor or genetic deletion markedly suppressed the late-phase PARP-1 activation induced by H2O2, suggesting that JNK1 contributes to the sustained activation of PARP-1. Such findings were supported by the temporal pattern of nuclear translocation of activated JNK and a direct protein–protein interaction between JNK1 and PARP-1 in H2O2-treated cells. Finally, in vitro kinase assay suggests that PARP-1 may serve as the direct phosphorylation target for JNK1. Taken together, data from our study reveal a novel underlying mechanism in H2O2-induced nonapoptotic cell death: JNK1 promotes a sustained PARP-1 activation via nuclear translocation, protein–protein interaction and PARP-1 phosphorylation.
Similar content being viewed by others

Main
Reactive oxygen species (ROS) refers to a group of oxygen-centered free radicals such as superoxide anion, hydroxyl radicals and hydrogen peroxide (H2O2).1 The production of these free radicals, which can be as high as millimolar quantities in the production sites, has been suggested to be involved in many physiological and pathological conditions.2 It is known that the disturbed cell redox status owing to enhanced level of ROS and/or impaired antioxidant defense mechanisms induces a cellular oxidative stress, leading to various biological consequences, including apoptotic or necrotic cell death.3, 4 The involvement of ROS or oxidative stress in cell death is two-fold. First, ROS act as cell death initiators through their direct damaging effect on various macromolecules including protein, DNA and lipid.5 Depending on the concentration or the cell type, intracellular generated ROS or exogenously applied ROS can cause either apoptosis or necrosis.6 Second, ROS function as intermediate signaling molecules in the cell death signaling pathways. One example is the role of ROS in tumor necrosis factor (TNF)-mediated both apoptotic and necrotic cell death.7, 8
Poly(ADP-ribose) polymerase 1 (PARP-1) is the founding member of the PARP family, a group of nuclear enzymes functioning as a critical post-translational regulatory mechanism in the process of DNA damage repair via PARP-1-mediated poly(ADP-ribosyl)ation.9 Despite its beneficial effect in DNA repair, PARP-1 activation has been implicated in the cell death process. In the case of severe DNA damage, massive PARP-1 activation would quickly deplete cellular β-nicotinamide adenine dinucleotide (NAD+), leading to the failure in ATP production and eventually necrotic cell death.10, 11 Recently, PARP-1 has also been demonstrated to mediate caspase-independent cell death via apoptosis-inducing factor (AIF): the PARP-1 overactivation prompts mitochondrial dysfunction and the translocation of AIF from mitochondria to nuclei in cells treated with DNA-damaging agents.12
c-Jun N-terminal protein kinase (JNK) is an important member of the mitogen-activated protein (MAP) kinase family that plays critical physiological and pathological roles in cells.13 It has been well established that a variety of extracellular stimuli such as oxidative stress, UV and cytokines activate JNK via a MAP kinase cascade consisting of MAP kinase kinases (MAPKK) and MAP kinase kinase kinases (MAPKKK). The MAPKKK responsible for JNK activation are mainly MEKK1 and ASK1, which phosphorylate and activate two MAPKK, JNKK1/MKK4/SEK1 and JNKK2/MKK7.13 Several lines of evidence suggests that JNK is an important mediator in oxidative stress-induced apoptotic cell death, and the proapoptotic function of JNK has been demonstrated in various cell types stimulated with different forms of ROS.14, 15 On the other hand, the regulatory role of JNK in ROS-induced nonapoptotic cell death was less studied. One of our recent reports demonstrated that JNK plays a critical role in H2O2-induced caspase-independent nonapoptotic cell death in mouse embryonic fibroblasts (MEF) downstream of tumor necrosis factor receptor (TNFR)-associated factor 2 (TRAF2 and receptor-interacting protein (RIP).16 However, the signaling mechanism downstream of JNK activation in ROS-induced nonapoptotic cell death has not been fully elucidated. In this study, we demonstrate a novel regulatory role of JNK1 in H2O2-induced PARP-1 activation and subsequent nonapoptotic cell death. By nuclear translocation, direct protein–protein interaction and phosphorylation, JNK1 facilitates a sustained PARP-1 activation, leading to a severe depletion of cellular ATP, which consequently results in a nonapoptotic cell death.
Results
H2O2 induces a caspase-independent nonapoptotic cell death in wild-type MEF cells
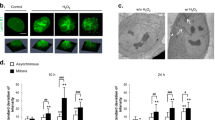
The form of cell death induced by H2O2 varies depending on the concentration as well as cell type.6 In this study, we first observed a dose-dependent increase in cell death in wild-type (wt) MEF treated with a range of H2O2 (up to 1000 μ M) and time-dependent increase in cell death induced by H2O2 (Figure 1a). Notably, the morphological features of cells treated with H2O2 were markedly different from those treated with TNFα. When typical apoptotic alterations such as cell blebbing and chromatin condensation were clearly observed in TNFα-treated cells, no similar changes were found in those cells treated with H2O2 (Figure 1b). Instead, many cells lost the membrane integrity (demonstrated by red nucleus stained with ethidium bromide (EB) at the early stage of H2O2 treatment, suggesting that those cells most probably underwent necrotic cell death. As caspase-3 is the principal effector caspase responsible for most of the apoptotic morphological changes, we further tested the involvement of caspase-3 activation in H2O2-induced cell death. No caspase-3 cleavage was detected in MEF cells treated with H2O2, whereas a complete cleavage was observed in cells treated with TNFα (Figure 1c). Consistently, two general caspase inhibitors (Z-VAD and Boc-D) failed to inhibit H2O2-induced cell death examined by a general cytotoxicity parameter, lactate dehydrogenase (LDH) leakage (Figure 1d). The effectiveness of those caspase inhibitors were confirmed by their ability to block TNFα-induced apoptosis in HeLa cells (data not shown). Data from this part of our study collectively demonstrate that H2O2-induced cell death is likely to be nonapoptotic/necrotic and independent of caspase-3 activation.

H2O2-induced caspase-independent nonapoptotic cell death in wt MEF cells. (a) Concentration and time-dependent cell death induced by H2O2 in wt MEF cells. Left panel: cells were treated with a range of H2O2 for 24 h; right panel: cells were treated with H2O2 (500 μ M) for various time. (b) Cell morphological changes detected byAO/EB staining. wt MEF cells were treated with either H2O2 (500 μ M) or TNFα (25 ng/ml+CHX 10 μg/ml) for 12 h followed by AO/EB staining (mixture of 100 μg/ml each of AO and EB) for 10 min. Then, the cells were examined and photographed using a fluorescence microscope. (c) Caspase-3 cleavage detected by WB. wt MEF cells were treated with H2O2 (500 μM × 12 h) or TNFα (25 ng/Ml) plus CHX (10 μg/ml) for 12 h. (d) General caspase inhibitors (z-VAD and Boc-D) were unable to block H2O2-induced cell death. Cells were pretreated with both inhibitors (50 μM) for 30 min before H2O2 exposure (500 μM × 24 h). In (a) and (d), the percentage of cell death was determined by LDH leakage and data were presented as means ± S.D. of three independent experiments (**P<0.01, t-test). In (b) and (c), representative results were presented from three independent experiments
H2O2-induced nonapoptotic cell death is dependent on PARP activation
PARP-1 is one of the main defense machinery against DNA damage including oxidative stress-induced DNA damage.11, 17, 18 Recently, a number of studies have suggested the involvement of PARP-1 activation in caspase-independent cell death induced by DNA damage agents or oxidation agents.12, 19 In this study, we first examined H2O2-induced PARP-1 activation in wt MEF. As shown in Figure 2a, a strong formation of poly (ADP-ribose) (PAR) polymer, a direct result of PARP-1 activation, was observed as early as 15 min after H2O2 exposure, whereas no PAR formation was detected in PARP-1 knockout MEF (PARP-1−/−). Moreover, the PARP-1−/− cells appeared to be largely resistant to H2O2-induced cell death (Figure 2b). Consistently, pretreatment with a specific PAPR inhibitor, 3AB, significantly inhibited formation of PAR polymer as well as the cellular ATP depletion (Figure 2c and d), and subsequently inhibited H2O2-induced cell death in wt MEF cells (Figure 2e). These observations clearly suggest that PARP-1 is an important mediator in H2O2-induced nonapoptotic cell death. To verify the above finding, we examined the effect of PARP-1 protein reconstitution on the susceptibility of PARP-1−/− MEF to H2O2-induced cell death. A PARP-1 expression vector20 was transiently transfected into the PARP-1−/−MEF cells. As shown in Figure 2f, PARP-1 reconstitution markedly restored the sensitivity to H2O2-induced cell death, thus further confirming the critical role of PARP-1 in H2O2-induced cell death.

PARP-1 activation and ATP depletion in H2O2-induced cell death. (a) Time course of PARP activation induced by H2O2 in wt MEF cells and PARP-1−/− cells. Cells were treated with H2O2 (500 μM) up to 60 min. The formation of PAR polymer was detected by WB. (b) H2O2-induced cell death in PARP-1−/− cells. wt MEF and PARP-1−/− MEF cells (PAPR-1−/−) were treated with H2O2 (500 μM) for 24 h. Upper panel: representative image showing the morphological changes of MEF cells after H2O2 treatment; lower panel: quantification of time-dependent cell death of wt MEF and PARP-1−/− MEFs by LDH leakage. (**:comparing with PARP-1−/− MEF group) (c) Effect of 3AB, a specific PARP inhibitor, on H2O2-induced PARP-1 activation, as detected by WB for the formation of PAR polymer. wt MEF cells were pretreated with 3AB (2 mM × 30 min) before H2O2 exposure (500 μM × 30 min). (d) Effect of 3AB on H2O2-induced intracellular ATP depletion. wt MEF cell were treated the same as in panel (c). The cells were typsinized and resuspended in PBS, and then subjected to cellular ATP detection as described in materials and methods. (e) Effect of 3AB on H2O2-induced cell death. Cells were pretreated with 3AB (2 mM × 30 min) before H2O2 exposure (500 μM × 12 h). The percentage of cell death was detected and quantified by LDH leakage. (f) Reconstitution of PARP-1 protein restores the susceptibility of PARP-1 −/− cell to H2O2-induced cell death. PARP-1−/− cells were transiently transfected with PARP-1 expression vector or pcDNA3 control vector. Twenty-four hours after transfection, cells were treated with varied concentrations of H2O2 (up to 1000 μM × 24 h). Upper panel: the reconstitution of PARP-1 protein was confirmed by WB; lower panel: quantification of dose-dependent cell death in PARP-1−/− MEF transfected with either pcDNA3 or PARP-1 expression vector, using a method same as in Figure 2b. All the numeric data were presented as means±S.D. of three independent experiments (**P<0.01, comparing with pcDNA group, Student's t-test)
JNK regulates PARP activation in H2O2-induced cell death
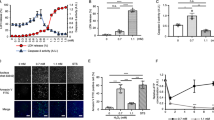
JNK is one of the main stress-responsive kinases and has been implicated in oxidative stress-induced apoptosis.21 In our previous study, we have shown that JNK, especially the JNK1 activation, has a critical role in promoting H2O2-induced nonapoptotic cell death.16 As both PARP-1 and JNK1 play an indispensable role in H2O2-induced cell death, here we try to further examine the possible functional inter-relationship between JNK and PAPR activation in H2O2-induced cell death. As shown in Figure 3a, significant JNK activation started 15–30 min after H2O2 exposure and maintained at the peak level throughout the period of treatment (up to 2 h), as detected by kinase assay as well as by Western blot (WB) for phosphor-JNK. When pretreated, the wt MEF cells with either a JNK inhibitor (SP600125) or a PARP inhibitor (3AB), SP600125 could effectively inhibit H2O2-induced JNK activation, whereas PARP inhibitor 3AB has no effect on H2O2-induced JNK activation (Figure 3b). Moreover, there was no discernible decrease in JNK activation in PARP-1−/− cells in comparison with wt MEF, suggesting that PARP-1 may not be important for H2O2-induced JNK activation (Figure 3c). In contrast, both 3AB and SP600125 significantly suppressed PARP-1 activation (Figure 3d). These findings thus suggest that JNK activation is likely to act as an important regulator upstream of PARP activation in H2O2-treated cells.

Effects of SP600125 and 3AB on JNK activation of PARP-1 activation. (a) H2O2-induced JNK activation detected by JNK kinase assay (KA) and WB. wt MEF cells were treated with H2O2 (500 μM) for up to 120 min. Total JNK was examined using WB as the loading control. (b) Effects of SP600125 and 3AB on H2O2-induced JNK activation. wt MEF cells were pretreated with either SP600125 (20 μM) or 3AB (2 mM) for 30 min before H2O2 exposure (500 μM × 30 min). JNK activation was determined by JNK kinase assay. (c) JNK activation in PARP-1−/− cells. wt MEF and PARP-1−/− cells were treated with H2O2 (500 μM × 30 min) and then subjected to WB for JNK activation. (d) Effects of SP600125 and 3AB on H2O2-induced PARP-1 activation. wt MEF cells were pretreated with either SP600125 (20 μM) or 3AB (2 mM) for 30 min before H2O2 exposure (500 μM 230 min). Cells were collected and subjected to WB. PAPR-1 activation was evaluated by detection of the formation of PAR polymer. All the kinase assay and WB data were representative results from three independent experiments
JNK1 plays a positive regulatory role in promoting a sustained PARP activation
It is generally believed that the PAPR activation is a fast responsive process, which can be observed within minutes of exposure to DNA-damaging agents.9 Under H2O2 treatment, there is a rapid PARP activation, reaching the peak level at 15 min (Figures 2a and 4). However, JNK activation appeared to be slower and only reached the peak level after 30 min (Figure 3a). Such a temporal difference between PARP and JNK activation thus raises an interesting question: how does JNK affect the pattern of PAPR activation in response to H2O2 treatment? To elucidate this question, we examined H2O2-induced PARP activation in an extended period up to 4 h. It was interesting to note that the inhibition of JNK activation by SP600125 pretreatments significantly shortened the duration of PARP activation, without significant effect on the early time point (15 min) (Figure 4a). It thus seems that there are two possible phases of PARP activation in response to H2O2, and JNK1 may positively regulate PARP activation in the second and extended phase. This notion was further supported by using JNK1 knockout MEF cells (JNK1−/−). First, H2O2-activated PARP-1 was largely diminished in JNK1−/− cells, although it was still visible at the early stage of H2O2 treatment (15 min) (Figure 4b). It is also noted that the phosphorylation of total JNK could still be detected in the JNK1−/− cells owing to the presence of JNK2, which suggests that activation of JNK1, but not JNK2, contributes to the sustained phase of PARP activation. In order to verify the role of JNK1 in H2O2-induced PARP activation, we examined the effect of JNK1 protein reconstitution on H2O2-induced PARP-1 activation, using JNK1−/− cells transfected with a JNK1 expression vector (HA-JNK1). As shown in Figure 4c, the reconstitution of JNK1 induced a more extensive and sustained PARP activation upon H2O2 exposure, well corresponding to the sustained JNK activation observed in those cells (Figure 4c). Therefore, the above data collectively indicate that JNK1 may play a positive role in promoting sustained PARP activation.

The regulatory role of JNK1 in promoting sustained PARP-1 activation. (a) Transient PARP-1 activation in H2O2-treated cells in the presence of SP600125. wt MEF cells were pretreated with SP600125 (20 μM) for 30 min, followed by treatment with H2O2 (500 μM) for up to 4 h. PARP-1 and JNK activation was determined by WB as described above. (b) H2O2-induced PARP activation in JNK1−/− cells. wt MEF and JNK1−/− MEF cells were treated with H2O2 (500 μM) for up to 4 h. PARP-1 and JNK activation was determined by WB. (c) Restoration of PARP activation by reconstitution of JNK1 in JNK1−/− cells. JNK1−/− cells reconstituted with JNK1 were treated with H2O2 (500 μM up to 4 h) and then subjected to WB. All the WB data were representative blots from at least two independent experiments
H2O2 induces JNK nuclear translocation upon its activation
JNK nuclear translocation is known to be one of its signal transduction mechanisms.22 To further understand the possible effects of JNK1 on PARP activation, here we examined the nuclear translocation of phospho-JNK upon H2O2 treatment in wt MEF cells, using immunofluoresence staining. As shown in Figure 5, there was a significant increase in the phosphor-JNK staining 15 min after H2O2 treatment. Moreover, a progressively increased nuclear phospho-JNK staining was found from 30–60 min. Such a temporal pattern may help to explain the earlier findings that JNK1 mainly affects PARP-1 activation at the later stage of H2O2 exposure. More importantly, as PARP is a nuclei-localized protein,23 the evident nuclear translocation of activated JNK provides a prerequisite for the functional interaction between JNK and PARP.

Nuclear translocation of activated JNK. wt MEF cells were treated with H2O2 (500 μM) for up to 60 min. At the end of the treatment, cells were fixed and the phospho-JNK was examined using immunostaining with counterstaining of nuclei by DAPI. All the images were captured by Olympus FLOVIEW V500 confocal microscope. Data were the representative images from three independent experiments
JNK1 directly associates and phosphorylates PAPR-1
It has been well established that components of nuclear transcription factor AP-1 are the most important nuclear substrates for JNK in stress response.24 Currently, there is no direct evidence demonstrating that JNK targets other nuclear proteins and regulates their functions. Here, we attempted to elucidate the direct function impact of JNK on PARP-1. We first examined the protein–protein interaction between JNK and PARP by endogenous co-immunoprecipitation (co-Ip) experiments. As shown in Figure 6a, H2O2 treatment clearly promoted the protein–protein interaction between JNK1 and PARP-1, evidenced by the increased amount of PARP-1 pulled down by JNK1 (top panel). Importantly, the temporal pattern of such interaction is found to be generally consistent with JNK1 activation (Figure 6a, second panel) and nuclear translocation (Figure 5). Moreover, the interaction between JNK1 and PARP-1 is believed to be specific as proliferating-cell nuclear antigen (PCNA), a DNA replication-related protein abundantly localized in nuclei, was not found in the complex (Figure 6a, bottom panel). All these observations clearly suggest that H2O2 is capable of inducing a direct association between JNK1 and PARP-1.

Direct association and phosphorylation of PARP-1 by JNK1. (a) Protein–protein interaction between PARP-1 and JNK1 upon H2O2 treatment. wt MEF cells were treated with H2O2 (500 μM) for up to 120 min. Cell lysate was immunoprecipitated with anti-JNK1 antibody. The interaction between PARP-1 and JNK1 was detected by WB. (b) Direct phosphorylation of PARP-1 by JNK1 in vitro. wt MEF cells were pretreated with/without SP600125 (20 μM) for 30 min followed by H2O2 (500 μM) treatment for up to 1 h. The activated JNK1 proteins were immunoprecipitated by anti-JNK1 antibody from total cell lysate and the phosphorylation of PARP-1 by immunoprecipitated JNK1 was detected by kinase assay. A known JNK substrate GST-c-Jun was used as a positive control. The cell lysate was equally divided and immunoprecipitated for the detection of phosphorylation of PARP-1 and GST-c-Jun, respectively. The Coomassie blue stain was carried out to validate the equal loading. (c) Phosphorylation of PARP-1 by recombinant JNK1 protein in vitro. Recombinant PARP-1 protein was incubated with active recombinant JNK1 protein for 30 min at 37°C and subjected to SDS-PAGE and autoradiography. The Coomassie blue stain was carried out to validate the equal loading. In panels (a–c), data presented were the representatives from three independent experiments. (d) An illustration showing the proposed effect of JNK1 on PARP-1 activation and subsequent cell death. The dotted line indicates the positive regulatory role of PARP-1 on JNK activation via RIP-TRAF2 as reported previously19
After establishing the protein–protein interaction between JNK1 and PARP-1, we next examined the possibility that PARP-1 serves as the substrate for JNK1. In this study, we performed the in vitro kinase assay by using human recombinant PARP-1 protein as the substrate.25 As shown in Figure 6b, phosphorylation of PARP-1 protein could be observed depending on time after incubation with immunoprecipitated JNK1 protein from H2O2-treated cells. The phosphorylation of GST-c-Jun, the known substrate of JNK, was also detected as a positive control. More importantly, a time-dependent change of 32P-labeled PARP-1 was well correlated to the temporal pattern of JNK1 activation, and the phosphorylation of PARP-1 was greatly suppressed depending on time by a JNK-inhibitor SP600125 (Figure 6b). To exclude the possibility that phosphorylation of PARP-1 observed in the above system was mediated by other proteins coexisting in the immunoprecipitation complex, we here used active recombinant JNK1 protein in the kinase assay. As show in Figure 6c, the presence of activated JNK1 produced both JNK1 auto-phosphorylation as well as a strong phosphorylation of GST-c-Jun. More importantly, consistent with the data in Figure 6b, the recombinant JNK1 protein also clearly phosphorylated PARP-1 protein. Hence, data from this part of our study provide convincing evidence that PARP-1 serves as a direct molecular target for JNK1 in cells under oxidative stress.
Discussion
The involvement of ROS or oxidative stress in apoptosis has been extensively studied, either as direct inducers or as intermediate regulators.6, 26 In contrast, relatively little is known about the signaling pathways in ROS or oxidative stress-mediated nonapoptotic cell death. An earlier report from our laboratory demonstrated a unique signaling pathway in H2O2-induced caspase-independent non-apoptotic cell death: H2O2 prompts protein–protein interaction between RIP and TRAF2 that subsequently mediates JNK activation.16 In this study, we attempted to elucidate the molecular mechanisms downstream of JNK in H2O2-mediated nonapoptotic cell death. Here, by using both pharmacological and genetic approaches, we identified PARP-1 as an important downstream target of JNK in H2O2-induced nonapoptotic cell death. By nuclear translocation, direct interaction and phosphorylation of PAPR-1, activated JNK1 acts as a positive regulator to render a sustained PARP-1 activation and subsequent cell death. Hence, results from this study reveal a novel regulatory mechanism by JNK1 in controlling the non-apoptotic cell death processes under oxidative stress.
JNK is one of the main groups of MAPK that are readily activated in response to various environmental stresses and plays a critical role in the processes deciding the fate of the cells.13 Although most of the current evidence points to the proapoptotic function of JNK in apoptosis induced by ROS or oxidative stress via sustained JNK activation,27 there are reports showing the importance of ROS-mediated JNK activation in non-apoptotic or necrotic cell death. For instance, ROS-mediated sustained JNK activation plays a critical role in TNFα-induced necrotic cell death.28, 29 In our previous study, we have demonstrated that JNK activation is closely involved in nonapoptotic cell death elicited by exogenously applied H2O2 in MEF cells.16 However, the molecular events downstream of JNK in nonapoptotic or necrotic cell death are still elusive.
PARP-1 is a highly abundant nuclear protein and plays a critical role in DNA repair, DNA stability and gene transcription.30 Several lines of evidence have suggested that PARP-1 is the key executor involved in necrotic cell death induced by ROS or DNA-damaging agents: excessive activation of PARP-1 leads to depletion of NAD+ and collapse of ATP production, resulting in necrotic cell death as a consequence of energy loss.18 Recent studies have also highlighted some other mechanisms downstream of PARP-1 activation in cell death, including transcriptional regulation of cell death related genes, such as nuclear factor-êB and p5331 and induction of mitochondrial dysfunction and release of AIF to mediate caspase-independent cell death.12 In this study, we first confirmed the critical role of PARP-1 in H2O2-induced nonapoptotic cell death by the following observations: (i) resistance of PARP-1−/− MEF cells to H2O2-induced cell death (Figure 2), (ii) protective effect of 3AB, a PARP-1 inhibitor, on H2O2-induced cell death (Figure 3e) and (iii) restoration of susceptibility of PARP-1−/− MEF cells to H2O2-induced cell death by PARP-1 protein reconstitution. At present, relatively little is known about the upstream regulatory mechanisms contributing to PARP-1 activation. A recent study demonstrated a positive regulatory role of extracellular signal-regulated kinase 1/2 (ERK1/2) in N-methyl-N′-nitro-N-nitrosoguanidine (MNNG)-induced PARP-1 activation and cell death.25 In this study, we provide evidence that JNK may act as an upstream regulator of PARP activation in H2O2-induced cell death, based on observations using a JNK inhibitor and JNK1−/− MEF cells (Figures 3 and 4). More importantly, JNK1 is believed to act as a positive-modulator to promote a sustained PARP-1 activation, based on observations that JNK1 inhibition did not affect the initial PARP-1 activation, but markedly suppress PARP-1 activation in the extended period. Such a notion is further supported by the temporal pattern of JNK1 nuclear translocation upon H2O2 exposure (Figure 5). Another interesting finding is that in JNK1−/− cells, there is still considerable level of total JNK activation upon H2O2 treatment, due to the activation of JNK2. It is thus believed that JNK1, but not JNK2, play a dominant role in regulating PARP-1 activity in response to H2O2 exposure. Such a finding is in line with our previous observation that only JNK1−/−, but not JNK2−/− MEF, is resistant to H2O2-induced cell death.16
The next important question needs to be answered is how PARP-1 activity is regulated by JNK1. Currently, several regulatory mechanisms of PARP-1 activity have been proposed: first, PARP auto-poly(ADP-ribosyl)ation as a negative model of modulation of PARP-1 activity and32 second, phosphorylation of PARP-1 by a number of protein kinases, such as protein kinase C (PKC), DNA-dependent protein kinase (DNA-PK), AMP-activated protein kinase (AMPK), and ERK1/2.25, 33, 34 In this study, we, for the first time, provide evidence showing that PARP-1 may serve as a substrate for JNK1. First, PARP-1 interacts with JNK1 in H2O2-treated cells detected by the endogenous co-immunoprecipitation, with a temporal pattern largely consistent with JNK activation and nuclear translocation. Second, either activated JNK1 immunoprecipitated from H2O2-treated cells or the active recombinant JNK1 was able to directly phosphorylate recombinant PARP-1 protein in the in vitro kinase assay.
At this stage, it is still controversial whether PARP-1 protein phosphorylation promotes or suppresses its activity. For example, PKC or DNA-PK has been shown to phosphorylates and inhibits PAPR-1 activity by disrupting its DNA-binding capability.33, 35 In contrast, PARP-1 phosphorylation mediated by AMPK or ERK1/2 was found to promote the catalytic activity of PARP-1.25, 36 The contradictory role of phosphorylation suggests that the impact of protein phosphorylation status on the bioactivity of PARP-1 is context-dependent. Data from this study tend to suggest that JNK1 positively regulates PARP-1 activity by means of phosphorylation, similar to the function of ERK1/2 on PARP-1 in MNNG-treated cells.25 Nevertheless, the exact nature of such effect remains to be further investigated, including the identification of the phosphorylation sites and the physiological relevance of phosphorylation of PARP-1 by JNK1.
Interestingly, our finding that JNK acts as an upstream regulator of PARP-1 is complimentary to some recent reports in which PARP-1 is found to contribute to JNK activation. For instance, Xu et al. demonstrated that, in cells treated with a DNA damage agent MNNG, PARP-1 contributes to JNK activation and cell death via RIP and TRAF2.19 Similarly, PARP-1 has been found to modulate AP-1 transcriptional activity through JNK activation.37 It is thus possible that there is a positive feedback loop between JNK and PARP-1 that affects each other's function depending on the nature of stimuli and cell type (Figure 6d).
Taken together, results from our study reveal a novel signaling pathway underlying ROS or oxidative stress-mediated nonapoptotic/necrotic cell death: H2O2-activated JNK1 acts a positive regulator in promoting a sustained PARP-1 activation via phosphorylation. Understanding such a mechanism helps to appreciate the molecular events controlling ROS-mediated nonapoptotic cell death downstream of JNK.
Materials and Methods
Reagents
H2O2, cycloheximide (CHX), 3-amino benzamide (3AB), acridine orange (AO), EB, ATP detection kit, cell dissociation solution and human recombinant PARP-1 protein were purchased from Sigma (St. Louis, MO, USA). The specific JNK inhibitor SP600125 and general caspase inhibitors Z-VAD and Boc-D were from Calbiochem (San Diego, CA, USA). Anti-phospho-JNK and anti-caspase-3 antibodies were purchased from Cell Signaling (Danvers, MA, USA). Anti-PAPR-1 and anti-JNK1 antibody for immunoprecipitation and anti-PCNA for WB were from Santa Cruz (Santa Cruz, CA, USA). Anti-PAR, anti-JNK1 and anti-PARP antibody were from BD Pharmingen (Los Angeles, CA, USA). Mouse recombinant TNFα was from R&D (Minneapolis, MN, USA). The general cytotoxicity detection (LDH) kit was from Roche (Indianpolis, IN, USA). Active recombinant JNK1 protein was from Upstate (Lake Placid, NY, USA).
Cell culture and detection of cell death
Immortalized wt and JNK1 knockout (JNK1−/−) MEF were cultured in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum, glutamine (2 mM), penicillin (100 U/ml) and streptomycin (100 μg/ml). PARP-1 knockout (PARP-1−/−) and its corresponding wt MEF were obtained from Dr. ZQ Wang38 and cultured similarly as above. Cells were subcultured 12 h before treatment and reached about 70% cell confluence at the time of treatment. Upon various designated treatments, cell death in MEF was examined using the following methods: (i) AO/EB staining to distinguish apoptotic and necrotic cell death in morphology39 and (ii) LDH leakage quantified using a cytotoxicity detection kit (Roche) for measuring the changes of plasma membrane permeability.
ATP measurement
Intracellular ATP level was determined using a bioluminescent somatic cell assay kit (Sigma) based on the procedures recommended by the manufacturer. Briefly, after designated treatments, cells culture dishes were immediately put on ice, and cells were collected by ice-cold cell dissociation solution (Sigma) and then resuspended in ice cold phosphate buffer saline (PBS) (pH=7.8). About 100 000 cells were incubated with freshly prepared ATP assay mix and ATP releasing reagent and then subjected to bioluminescent detection. The ATP level was presented as percentage to the untreated control group.
Transient transfection
PARP-1 expression vector (pPARP31) was kindly provided by Professor Alexander Bürkle.20 The transient transfection of PARP-1 expression vector into PARP-1−/− cells were performed using electroporation-based method. The vector (4 μg DNA) was diluted with MEF2 nucleofector reagent (Amaxa GmbH, Koeln, Germany) and mixed with 1 × 106 cells. After incubation for 5 min, cells were subjected to electroporation using Program A23. The transfection efficiency was examined by transfection MEF cells with pMaxFP™-Green vector (Amaxa) in parallel. After 24 h, more than 80% cells were confirmed to be successfully transfected. The transient transfection of HA-JNK1 expression vector into JNK1−/− cells was also carried out as described above.
JNK kinase assay and detection of phosphorylation of recombinant PARP-1
JNK kinase assay was performed as described previously.16 Briefly, at the end of designated experiments, cell lysate was collected using M2 buffer (20 mM Tris at pH 7, 0.5% NP-40, 250 mM NaCl, 3 mM EDTA, 3 mM EGTA, 2 mM DTT, 0.5 mM PMSF, 20 mM -glycerol phosphate, 1 mM sodium vanadate, 1 μg/ml leupeptin). JNK1 protein was immunoprecipitated from 500 μg of total cell lysate with 1 μg anti-JNK1 antibody (Santa Cruz) and harvested with protein A-sepharose beads (Roche). The immunoprecipitates were washed extensively with M2 buffer for three times and incubated with GST-c-Jun in complete kinase assay buffer (20 mM HEPES pH 7.5, 20 mM β-glycerol phosphate, 10 mM MgCl2, 1 mM DTT, 10 mM PNPP, 50 μ M sodium vanadate, 20 μ M ATP) by the addition of 32P-γ-ATP. After incubation at 30°C for 30 min, the reaction was stopped by the addition of SDS sample buffer. The samples were then separated on a SDS-PAGE and visualized by autoradiography.
In vitro detection of phosphorylation of recombinant PARP-1 was performed generally similar to JNK kinase assay. For the assay using immunoprecipitated JNK1, the cell lysate was collected using M2 buffer at the end of the designated treatment. After balanced by total protein content, the JNK1 protein was immunoprecipitated with anti-JNK1 antibody (Santa Cruz) and protein A-sepharose beads. Then, the assay was performed for 30 min at 30°C with 32P-γ-ATP and recombinant PAPR-1 protein (5 U) as a substrate. GST-c-Jun was also included in parallel as a positive control for JNK-1 activity. For the assay using active recombinant JNK1 protein, 2 μg of activated recombinant JNK1 protein was incubated with recombinant PAPR-1 protein (5 U) directly at 30°C for 30 min with 32P-γ-ATP in complete kinase buffer and then subjected to SDS-PAGE and autoradiography.
Immunofluorescence and confocal microscopy
Briefly, the wt MEF cells were plated on the eight well chamber slides (Nunc) and cultured overnight before treatments. At the end of designated treatments, cells were washed with PBS once and fixed in 3.75% paraformaldehyde for 1 h at room temperature and permeabilized with 0.1% Triton-X in PBS for 5 min and blocked in blocking solution (2% BSA, 0.2% tween-20 in PBS) for another 1 h. Cells were further incubated with anti-phospho-JNK antibody (Cell signaling) overnight at 4°C with gentle shaking. After washing with washing buffer (PBS with 0.2% Tween-20) for three times, cells were further incubated with fluorescein isothiocyanate-labeled secondary antibody (1:100) for 1 h at room temperature. Then the slides were extensively washed with washing buffer and amounted with ProLong® Gold Anti-Fade mounting solution with DAPI (Invitrogen). The immunofluorescence was visualized by Olympus FLOVIEW V500 confocal microscope.
Co-IP and Western blot
For the Co-IP experiments, cell lysate was prepared as described above using M2 buffer. The cell lysate was mixed and incubated with anti-JNK1 antibody (Santa Cruz) and protein A-sepharose beads (Roche) and incubated at 4°C in a head-to-head rotator for at least 4 h. After extensive washing with the M2 cell lysis buffer for four times, protein A beads were boiled in SDS-sample buffer to release the protein. Then, the bound proteins were resolved on SDS-PAGE and probed with respective antibodies in WB.
For WB, after various designated treatments, cells were collected and washed with PBS before they were lysed in M2 buffer. The cell lysate was collected after centrifugation (12 000 r.p.m. × 15 min) and 30 μg of protein was then fractionated on SDS-PAGE and blotted onto polyvinylidene-fluoride membrane (Bio-Rad). After blocking in 5% milk in PBST (PBS with 0.05% Tween 20) for 1 h, the membrane was probed with various first antibodies overnight at 4°C, followed by incubation with respective second antibodies for 1 h. The membrane was washed in PBST and developed with enhanced chemiluminescence following the manufacturer's instructions (Pierce).
Abbreviations
- ROS:
-
reactive oxygen species
- H2O2:
-
hydrogen peroxide
- JNK:
-
c-Jun N-terminal kinase
- PARP-1:
-
poly (ADP-ribose) polymerase-1
- PAR:
-
poly (ADP-ribose)
- NAD+:
-
β-nicotinamide adenine dinucleotide
- AIF:
-
apoptosis-inducing factor
- RIP:
-
receptor-interacting protein
- TRAF2:
-
tumor necrosis factor receptor (TNFR)-associated factor 2
- 3AB:
-
3amino benzamide
- AO:
-
acridine orange
- EB:
-
ethidium bromide
- MNNG:
-
N-methyl-N′-nitro-N-nitrosoguanidine
References
Sies H . Oxidative stress: oxidants and antioxidants. Exp Physiol 1997; 82: 291–295.
Rhee SG . Cell signaling H2O2 a necessary evil for cell signaling. Science 2006; 312: 1882–1883.
Kwon YW, Masutani H, Nakamura H, Ishii Y, Yodoi J . Redox regulation of cell growth and cell death. Biol Chem 2003; 384: 991–996.
Hampton MB, Orrenius S . Redox regulation of apoptotic cell death. Biofactors 1998; 8: 1–5.
Tang DG, La E, Kern J, Kehrer JP . Fatty acid oxidation and signaling in apoptosis. Biol Chem 2002; 383: 425–442.
Gardner AM, Xu FH, Fady C, Jacoby FJ, Duffey DC, Tu Y et al. Apoptotic vs. nonapoptotic cytotoxicity induced by hydrogen peroxide. Free Radic Biol Med 1997; 22: 73–83.
Shen HM, Pervaiz S . TNF receptor superfamily-induced cell death: redox-dependent execution. FASEB J 2006; 20: 1589–1598.
Garg AK, Aggarwal BB . Reactive oxygen intermediates in TNF signaling. Mol Immunol 2002; 39: 509–517.
Virag L . Structure and function of poly(ADP-ribose) polymerase-1: role in oxidative stress-related pathologies. Curr Vasc Pharmacol 2005; 3: 209–214.
Herceg Z, Wang ZQ . Failure of poly(ADP-ribose) polymerase cleavage by caspases leads to induction of necrosis and enhanced apoptosis. Mol Cell Biol 1999; 19: 5124–5133.
Koh DW, Dawson TM, Dawson VL . Mediation of cell death by poly(ADP-ribose) polymerase-1. Pharmacol Res 2005; 52: 5–14.
Yu SW, Wang H, Poitras MF, Coombs C, Bowers WJ, Federoff HJ et al. Mediation of poly(ADP-ribose) polymerase-1-dependent cell death by apoptosis-inducing factor. Science 2002; 297: 259–263.
Johnson GL, Lapadat R . Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science 2002; 298: 1911–1912.
Lei K, Nimnual A, Zong WX, Kennedy NJ, Flavell RA, Thompson CB et al. The Bax subfamily of Bcl2-related proteins is essential for apoptotic signal transduction by c-Jun NH(2)-terminal kinase. Mol Cell Biol 2002; 22: 4929–4942.
Deng X, Xiao L, Lang W, Gao F, Ruvolo P, May Jr WS . Novel role for JNK as a stress-activated Bcl2 kinase. J Biol Chem 2001; 276: 23681–23688.
Shen HM, Lin Y, Choksi S, Tran J, Jin T, Chang L et al. Essential roles of receptor-interacting protein and TRAF2 in oxidative stress-induced cell death. Mol Cell Biol 2004; 24: 5914–5922.
Bouchard VJ, Rouleau M, Poirier GG . PARP-1, a determinant of cell survival in response to DNA damage. Exp Hematol 2003; 31: 446–454.
Ha HC, Snyder SH . Poly(ADP-ribose) polymerase is a mediator of necrotic cell death by ATP depletion. Proc Natl Acad Sci USA 1999; 96: 13978–13982.
Xu Y, Huang S, Liu ZG, Han J . Poly(ADP-ribose) polymerase-1 signaling to mitochondria in necrotic cell death requires RIP1/TRAF2-mediated JNK1 activation. J Biol Chem 2006; 281: 8788–8795.
Van Gool L, Meyer R, Tobiasch E, Cziepluch C, Jauniaux JC, Mincheva A et al. Overexpression of human poly(ADP-ribose) polymerase in transfected hamster cells leads to increased poly(ADP-ribosyl)ation and cellular sensitization to gamma irradiation. Eur J Biochem 1997; 244: 15–20.
Deng Y, Ren X, Yang L, Lin Y, Wu X A . JNK-dependent pathway is required for TNFalpha-induced apoptosis. Cell 2003; 115: 61–70.
Cavigelli M, Dolfi F, Claret FX, Karin M . Induction of c-fos expression through JNK-mediated TCF/Elk-1 phosphorylation. Embo J 1995; 14: 5957–5964.
Lindahl T, Satoh MS, Poirier GG, Klungland A . Post-translational modification of poly(ADP-ribose) polymerase induced by DNA strand breaks. Trends Biochem Sci 1995; 20: 405–411.
Leppa S, Bohmann D . Diverse functions of JNK signaling and c-Jun in stress response and apoptosis. Oncogene 1999; 18: 6158–6162.
Kauppinen TM, Chan WY, Suh SW, Wiggins AK, Huang EJ, Swanson RA . Direct phosphorylation and regulation of poly(ADP-ribose) polymerase-1 by extracellular signal-regulated kinases 1/2. Proc Natl Acad Sci USA 2006; 103: 7136–7141.
Nakano H, Nakajima A, Sakon-Komazawa S, Piao JH, Xue X, Okumura K . Reactive oxygen species mediate crosstalk between NF-kappaB and JNK. Cell Death Differ 2006; 13: 730–737.
Tobiume K, Matsuzawa A, Takahashi T, Nishitoh H, Morita K, Takeda K et al. ASK1 is required for sustained activations of JNK/p38 MAP kinases and apoptosis. EMBO Rep 2001; 2: 222–228.
Kamata H, Honda S, Maeda S, Chang L, Hirata H, Karin M . Reactive oxygen species promote TNFalpha-induced death and sustained JNK activation by inhibiting MAP kinase phosphatases. Cell 2005; 120: 649–661.
Lin Y, Choksi S, Shen HM, Yang QF, Hur GM, Kim YS et al. Tumor necrosis factor-induced nonapoptotic cell death requires receptor-interacting protein-mediated cellular reactive oxygen species accumulation. J Biol Chem 2004; 279: 10822–10828.
Kim MY, Zhang T, Kraus WL . Poly(ADP-ribosyl)ation by PARP-1: ‘PAR-laying’ NAD+ into a nuclear signal. Genes Dev 2005; 19: 1951–1967.
Chiarugi A . Poly(ADP-ribose) polymerase: killer or conspirator? The ‘suicide hypothesis’ revisited. Trends Pharmacol Sci 2002; 23: 122–129.
Zahradka P, Ebisuzaki K . A shuttle mechanism for DNA-protein interactions. The regulation of poly(ADP-ribose) polymerase. Eur J Biochem 1982; 127: 579–585.
Ariumi Y, Masutani M, Copeland TD, Mimori T, Sugimura T, Shimotohno K et al. Suppression of the poly(ADP-ribose) polymerase activity by DNA-dependent protein kinase in vitro. Oncogene 1999; 18: 4616–4625.
Bauer PI, Farkas G, Buday L, Mikala G, Meszaros G, Kun E et al. Inhibition of DNA binding by the phosphorylation of poly ADP-ribose polymerase protein catalysed by protein kinase C. Biochem Biophys Res Commun 1992; 187: 730–736.
Tanaka Y, Koide SS, Yoshihara K, Kamiya T . Poly (ADP-ribose) synthetase is phosphorylated by protein kinase C in vitro. Biochem Biophys Res Commun 1987; 148: 709–717.
Walker JW, Jijon HB, Madsen KL . AMP-activated protein kinase is a positive regulator of poly(ADP-ribose) polymerase. Biochem Biophys Res Commun 2006; 342: 336–341.
Andreone TL, O'Connor M, Denenberg A, Hake PW, Zingarelli B . Poly(ADP-ribose) polymerase-1 regulates activation of activator protein-1 in murine fibroblasts. J Immunol 2003; 170: 2113–2120.
Wang ZQ, Auer B, Stingl L, Berghammer H, Haidacher D, Schweiger M et al. Mice lacking ADPRT and poly(ADP-ribosyl)ation develop normally but are susceptible to skin disease. Genes Dev 1995; 9: 509–520.
McGahon AJ, Martin SJ, Bissonnette RP, Mahboubi A, Shi Y, Mogil RJ et al. The end of the (cell) line: methods for the study of apoptosis in vitro. Methods Cell Biol 1995; 46: 153–185.
Acknowledgements
We thank Prof. Alexander Bürkle (German Cancer Research Center) for providing us the PARP-1 expression vector. We also thank Drs. GM Hur, S Choksi, Mr. YB Ong and Ms M Zhao for their technical support. This study was in part supported by a NCI oncology fellowship (to HM Shen) and research grants from the National University of Singapore (startup grant and provost matching grant to HM Shen).
Author information
Authors and Affiliations
Corresponding author
Additional information
Edited by H Ichijo
Rights and permissions
About this article
Cite this article
Zhang, S., Lin, Y., Kim, YS. et al. c-Jun N-terminal kinase mediates hydrogen peroxide-induced cell death via sustained poly(ADP-ribose) polymerase-1 activation. Cell Death Differ 14, 1001–1010 (2007). https://doi.org/10.1038/sj.cdd.4402088
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.cdd.4402088
Keywords
This article is cited by
-
CHK2 activation contributes to the development of oxaliplatin resistance in colorectal cancer
British Journal of Cancer (2022)
-
Interplay between ADP-ribosyltransferases and essential cell signaling pathways controls cellular responses
Cell Discovery (2021)
-
Marine alkaloid monanchoxymycalin C: a new specific activator of JNK1/2 kinase with anticancer properties
Scientific Reports (2020)
-
t-BuOOH induces ferroptosis in human and murine cell lines
Archives of Toxicology (2018)
-
MKP-1 suppresses PARP-1 degradation to mediate cisplatin resistance
Oncogene (2017)
