
Abstract
Here, we describe the non-redundant roles of caspase-activated DNase (CAD) and DNaseγ during apoptosis in the immature B-cell line WEHI-231. These cells induce DNA-ladder formation and nuclear fragmentation by activating CAD during cytotoxic drug-induced apoptosis. Moreover, these apoptotic manifestations are accompanied by inhibitor of CAD (ICAD) cleavage and are abrogated by the constitutive expression of a caspase-resistant ICAD mutant. No such nuclear changes occur during oxidative stress-induced necrosis, indicating that neither CAD nor DNaseγ functions under necrotic conditions. Interestingly, the DNA-ladder formation and nuclear fragmentation induced by B-cell receptor ligation occur in the absence of ICAD cleavage and are not significantly affected by the ICAD mutant. Both types of nuclear changes are preceded by the upregulation of DNaseγ expression and are strongly suppressed by 4-(4,6-dichloro-[1, 3, 5]-triazin-2-ylamino)-2-(6-hydroxy-3-oxo-3H-xanthen-9-yl)-benzoic acid (DR396), which is a specific inhibitor of DNaseγ. Our results suggest that DNaseγ provides an alternative mechanism for inducing nuclear changes when the working apoptotic cascade is unsuitable for CAD activation.
Similar content being viewed by others


Main
During B-lymphocyte development in the bone marrow, somatic gene rearrangements occur at the immunoglobulin (Ig) loci through V(D)J recombination.1 This produces a diverse repertoire of B cells, and allows the immune system to recognize a wide range of foreign antigens. However, creating such variety carries the risk of generating self-reactive B cells. To avoid autoimmune manifestations, self-reactive B cells can be rendered inactive (anergy), reconstituted in their antigen receptor (receptor editing) or eliminated by apoptosis (clonal deletion) at an immature stage of development.2 The clonal deletion of self-reactive B cells depends upon the sensitivity of their B-cell receptors (BCRs) to self-antigens. The strong engagement of surface IgM causes immature B cells to exit from the cell cycle and undergo apoptosis.3, 4 This cellular suicide program allows potentially harmful or redundant cells to be individually eliminated, in order to maintain healthy homeostasis in multicellular organisms.5 It is distinguished from necrosis by its specific morphologies (such as balloon-cell formation, chromatin condensation, and nuclear fragmentation) and biochemical markers (including the cleavage of death substrates by caspases and the degradation of genomic DNA into nucleosomal fragments).6, 7 The occurrence of nucleosomal DNA fragmentation is an important feature of apoptosis, and the presence of a ladder configuration in agarose gel electrophoresis provides reliable evidence of this process.7
Apoptotic signals are initiated by various physiological and pathological stimuli, and are transmitted by the successive activation of several protease cascades, including those of calpains, cathepsins, and caspases.8, 9 Although the utilization of apical proteases differs depending on the type of apoptotic stimulus, they subsequently promote the activation of downstream effector caspases (caspase-3, caspase-6, and caspase-7), which catalyze the proteolysis of death substrates (such as poly (ADP-ribose) polymerase-1 (PARP-1) and DNA-fragmentation factor 45 (DFF45)/inhibitor of caspase-activated DNase (ICAD)).8
DFF is a heterodimeric complex of DFF40 and DFF45, which is purified from the S100 fraction of HeLa cells on the basis of its activity to induce DNA-ladder formation in a cell-free apoptosis system.10 The murine homologs of DFF40 and DFF45 – caspase-activated DNase (CAD) and ICAD, respectively – have been isolated and characterized.11 CAD exists in living cells as an inactive complex with its natural inhibitor ICAD. Upon apoptosis induction, CAD is released from the complex by the caspase-3-mediated cleavage of ICAD, and catalyzes nucleosomal DNA fragmentation in dying cells.11 Thus, CAD is considered to have a major role in apoptosis accompanied by caspase-3 activation.
The existence of a nuclear apoptotic Ca2+/Mg2+-dependent DNase has long been suspected. We previously identified and purified the 33-kDa endonuclease DNaseγ.12 Sequence analysis of its complementary DNA (cDNA) confirmed it to be a novel member of the DNase I family.13 DNaseγ differs from the other family members by its ability to catalyze DNA fragmentation during apoptosis.14, 15 DNaseγ induction occurs in C2C12 cells during myogenic differentiation, and its suppression by the stable expression of its antisense RNA reduces DNA-ladder formation in spontaneous apoptosis during differentiation.16 It is also involved in spontaneous apoptosis during the neuronal differentiation of N1E-115 cells and the NGF-deprivation-induced apoptosis of neuronally differentiated PC12 cells.17 Thus, DNaseγ appears to play an important role in apoptosis during differentiation and specific developmental stages. As mentioned above, the apoptosis of self-reactive B cells occurs at a specific stage of B-cell development. Furthermore, DNaseγ is expressed at high levels in the bone marrow where clonal deletion takes place.14 Based on these facts, we proposed that the BCR-induced apoptosis of immature B cells might involve DNaseγ.
The current paper describes the specific expression of DNaseγ in immature B cells, and assesses its role in apoptotic DNA fragmentation induced by BCR cross-linking. We also report on the non-redundant roles of CAD and DNaseγ in apoptosis in the murine immature B-cell line WEHI-231.
Results
Expression profiles of CAD and DNaseγ in murine B cells
The expression levels of two apoptotic endonucleases, CAD and DNaseγ, were analyzed by Northern blots in several murine B-cell lines at various maturation stages. As shown in Figure 1a, CAD and ICAD were expressed at constant levels in the B cells. However, the DNaseγ expression level differed depending on the developmental stage. DNaseγ mRNA was detected at high levels in immature B cells (WEHI-231 and CH31). However, although lower DNaseγ expression levels were detected in the B-cell lines with mature phenotypes (WEHI-279 and A20), no specific hybridization was detected in pre-B cells (18–81) or plasma cells (J558L).

DNaseγ expression in murine B cells. (a) Northern blot analysis of DNaseγ, CAD, and ICAD mRNAs in mouse B-cell lines. Total RNAs isolated from the indicated cells were analyzed by Northern blots. The bands representing DNaseγ (2.2 kb), CAD (2.5 kb), and ICAD (2.3 kb) messenger RNAs (mRNAs) are indicated by arrowheads. The images of 28S and 18S ribosomal RNAs (rRNAs) are shown at the bottom for comparison. (b) B220/CD3 and IgM/IgD expression profiles of spleen B cells prepared from 5-Gy-irradiated (right panels) and non-irradiated (left panels) mice (as described in the Materials and Methods). The expression profiles of the indicated surface antigens were analyzed by FACScan using Cellquest software. (c) DNaseγ activities in mouse spleen B cells. DNaseγ activities in B-cell preparations, obtained from 5-Gy-irradiated (+) and non-irradiated (−) mice, were analyzed by an activity gel assay. The positions of the 33-kDa DNaseγ activities are indicated by the arrowhead. The image is representative of three independent experiments with similar results
The high DNaseγ expression level in immature B cells was confirmed in primary cells isolated from the spleen. Briefly, the mice were exposed to a sublethal dose of X-rays and Thy1.1-depleted spleen B cells were removed on day 13 post-irradiation. Approximately, 90% of the cells were positive for B220; the relative amounts of the immature phenotypes (IgMhi IgDlo) were 10 and 50% in the B-cell preparations obtained from the control and irradiated mice, respectively (Figure 1b). Figure 1c shows the results of the activity gel assay; the band representing DNaseγ activity was faint in the control preparation, whereas it was distinct in the preparation enriched with immature B cells.
These results revealed that the expression patterns of CAD and DNaseγ differed significantly during B-cell development: CAD was expressed at a constant level throughout B-cell ontogeny, whereas DNaseγ had a high expression level in immature B cells alone.
CAD is responsible for DNA and nuclear fragmentation during cytotoxic drug-induced apoptosis in WEHI-231 cells
The above-mentioned observations suggest that the utilization of CAD and DNaseγ is dependent upon the type of apoptotic stimulus in immature B cells. CAD appears to catalyze DNA fragmentation during apoptosis independent of the developmental stage, whereas DNaseγ is specifically involved in the apoptosis of immature B cells. The overexpression of a caspase-resistant form of ICAD selectively interferes with CAD activation without affecting DNaseγ activity.17 We therefore established WEHI-231 cells that were stably transfected with an expression vector for a caspase-resistant ICAD mutant (Figure 2a), and used them to define the contributions of CAD and DNaseγ in apoptotic DNA-ladder formation.

Role of CAD in chemical-induced apoptosis in WEHI-231 cells. (a) Stable expression of caspase-resistant ICAD in WEHI-231 cells. Western blot analyses of WEHI-231/Puro (Puro) and WEHI-231/ICAD-CR (ICAD-CR) cells. The bands representing the Myc and His-tagged ICAD-CR, and endogenous ICAD proteins, are indicated by an asterisk and an arrowhead, respectively. The result of the Western blot analysis of β-actin is given at the bottom for comparison. (b) Suppression of nucleosomal DNA fragmentation in WEHI-231/ICAD-CR cells treated with chemical apoptosis inducers. WEHI-231/Puro (Puro) and WEHI-231/ICAD-CR (ICAD-CR) cells were cultured for the indicated times in the presence of 0.1 μ M STS (left panels). WEHI-231/Puro (P) and WEHI-231/ICAD-CR (I) cells were cultured for 6 h in the absence (Cont) or presence of 100 μ M Etp, 3 μg/ml CHX, 1 μ M Tg, and 1 μ M Tm (right panels). DNAs prepared from the resulting cells were analyzed by 1.8% agarose gel electrophoresis to visualize DNA-ladder formation (upper panel), and proteins from the same cells were subjected to Western blot analysis to demonstrate the cleavage of ICAD (middle panel) and PARP-1 (lower panel). Closed arrowheads indicate the intact forms of PARP-1 (115 kDa) and ICAD (45 kDa). Open arrowheads show the bands of cleaved PARP-1 fragments (85 kDa). (c) Effects of suppression of CAD on STS-induced apoptotic morphological changes in WEHI-231 cells. WEHI-231/Puro (Puro) and WEHI-231/ICAD-CR (ICAD-CR) cells were cultured for 6 h in the absence (Control) or presence of 0.1 μM STS. The resulting cells were stained with Hoechst 33342 and observed by DIC or fluorescence microscopy (Hoechst). The black bar represents 50 μm
We initially examined the effects of CAD inhibition on apoptosis induced by cytotoxic drugs. Figure 2b shows the time course of apoptotic DNA fragmentation induced by staurosporine (STS) treatment. A nucleosomal DNA ladder was clearly produced in mock-transfected control cells (Puro), and this process was correlated with the disappearance of ICAD. By contrast, no ladder formation was observed in cells expressing the mutant ICAD (ICAD-CR). In both the control and ICAD-CR cells, the kinetics of PARP-1 cleavage were similar (Figure 2b). Thus, the overexpression of ICAD-CR was shown to suppress DNA fragmentation without affecting apoptotic death. Similar results were obtained with other cytotoxic drugs (etoposide (Etp), cycloheximide (CHX), thapsigargin (Tg), and tunicamycin (Tm)) (Figure 2b).
We also analyzed the effects of ICAD-CR on cellular and nuclear morphological changes (Figure 2c). Differential interference contrast (DIC) microscopy showed a balloon-like morphology, whereas the fluorescence field revealed extensive nuclear fragmentation in control cells treated with STS. Although the balloon-cell morphology was generally observed in ICAD-CR cells, the sequence of nuclear morphological changes was arrested at chromatin condensation and did not proceed to nuclear fragmentation. These findings indicate that CAD activation is required for both DNA-ladder formation and nuclear fragmentation during cytotoxic drug-induced apoptosis in WEHI-231 cells.
Neither CAD nor DNaseγ is activated during oxidative stress-induced necrosis in WEHI-231 cells
Oxidative stress caused by H2O2 induces both apoptosis and necrosis in a dose-dependent manner. We therefore examined the roles of CAD and DNaseγ in oxidative stress-induced apoptosis and necrosis in WEHI-231 cells. Low doses of H2O2 (∼50–100 μM) induced apoptotic DNA fragmentation and PARP-1 cleavage in the control cells (Figure 3a). The ladder formation was accompanied by ICAD cleavage, and was inhibited by ICAD-CR (Figure 3a). Dying cells retained their membrane integrity and exhibited similar morphological changes to those induced by STS (data not shown). These results indicate that H2O2 at doses up to 100 μM promotes CAD activation and apoptotic death in WEHI-231 cells; the cells undergo necrosis when treated with H2O2 at higher doses (∼0.3–1 mM). Microscopic analysis revealed necrotic characteristics, such as collapsed cellular morphologies, and an absence of nuclear changes (Figure 3b). The loss of membrane integrity was confirmed by a vital dye-exclusion test (data not shown). Under necrotic conditions, both the control and ICAD-CR cells died without showing DNA-ladder formation (Figure 3a). This suggests that neither CAD nor DNaseγ is activated during oxidative stress-induced necrosis in WEHI-231 cells.

Absence of DNA fragmentation and nuclear changes during oxidative stress-induced necrosis in WEHI-231 cells. (a) DNA-ladder formation induced by oxidative stress. WEHI-231/Puro (Puro) and WEHI-231/ICAD-CR (ICAD-CR) cells were cultured for 6 h in the presence of H2O2 at increasing doses. DNAs prepared from the resulting cells were analyzed by 1.8% agarose gel electrophoresis to visualize DNA-ladder formation (upper panel), and proteins from the same cells were subjected to Western blot analysis to demonstrate the cleavage of ICAD (middle panel) and PARP-1 (lower panel). Closed arrowheads indicate the intact forms of PARP-1 (115 kDa) and ICAD (45 kDa). Open arrowheads show the bands of cleaved PARP-1 fragments (85 kDa). The bands representing the Myc and His-tagged ICAD-CR are marked by an asterisk. (b) Morphological changes of necrotic WEHI-231 cells. WEHI-231/Puro (Puro) and WEHI-231/ICAD-CR (ICAD-CR) cells were cultured for 6 h in the presence of H2O2 at a necrotic dose (1 mM). The resulting cells were stained with Hoechst 33342 and observed by DIC or fluorescence microscopy (Hoechst). The black bar represents 50 μm
ICAD-CR fails to suppress DNA-ladder formation and nuclear fragmentation during BCR-mediated apoptosis in WEHI-231 cells
WEHI-231 cells have been widely used as a model to study the elimination mechanism of self-reactive B cells, because of their capacity to undergo apoptosis upon BCR stimulation.3, 4 Figure 4 shows the apoptotic features of WEHI-231 cells induced by BCR crosslinking. In contrast to apoptosis induced by cytotoxic drugs, nucleosomal DNA fragmentation was clearly observed in both the control (Puro) and ICAD-CR cells (Figure 4a). Although the ladder formation in the ICAD-CR cells was slightly impaired, it was less effective in suppressing BCR-mediated ladder formation than chemical apoptosis inducers. Consistent with this observation, ICAD-CR also failed to interfere with the appearance of fragmented nuclei during BCR-mediated apoptosis in WEHI-231 cells (Figure 4b). Among the cells showing apoptotic morphology, ∼76% of the control cells and 52% of the ICAD-CR cells exhibited nuclear fragmentation (Figure 4b).

Apoptosis of WEHI-231 cells induced by BCR ligation. (a) Apoptotic ladder formation induced by sIgM ligation. WEHI-231/Puro (Puro) and WEHI-231/ICAD-CR (ICAD-CR) cells were cultured for the indicated times in the presence of 10 μg/ml anti-IgM antibody. DNAs and proteins prepared from the resulting cells were subjected to 1.8% agarose gel electrophoresis (upper panel) and Western blot analysis for PARP-1 cleavage (lower panel), respectively. The bands representing intact (115 kDa) and cleaved (85 kDa) PARP-1 are indicated by closed and open arrowheads, respectively. (b) Apoptotic morphological changes induced by sIgM cross-linking. WEHI-231/Puro (Puro) and WEHI-231/ICAD-CR (ICAD-CR) cells were cultured for 36 h in the presence of 10 μg/ml anti-IgM antibody. The resulting cells were stained with Hoechst 33342 and observed by DIC or fluorescence microscopy (Hoechst). The insets indicate the relative numbers of nuclear-fragmented cells among the total cells showing balloon-like morphology. The relative number of total balloon-like cells is also given below each panel. All values are the mean±standard deviation (S.D.; n=4). The black bar represents 50 μm
To determine whether the activation of CAD occurred in response to BCR ligation, we performed Western blot analysis to detect the caspase-mediated cleavage of ICAD. As shown in Figure 5, apoptosis caused by both STS treatment and BCR cross-linking was accompanied by oligonucleosomal DNA fragmentation and PARP-1 cleavage. As expected, the disappearance of intact ICAD correlated well with the onset of DNA fragmentation in STS-induced apoptosis. By contrast, ICAD cleavage was barely observed during BCR ligation-induced apoptosis in WEHI-231 cells (Figure 5b). This explained the observation that ICAD-CR expression failed to suppress DNA fragmentation and nuclear morphological changes during apoptosis induced by BCR ligation. These results indicate that CAD does not play a major role in BCR-mediated apoptosis in WEHI-231 cells.

Absence of ICAD cleavage during BCR ligation-induced apoptosis in WEHI-231 cells. WEHI-231 cells were treated with 0.1 mM STS (STS) or 10 μg/ml anti-IgM antibody for the indicated time. The resulting cells were collected and subjected to (a) DNA electrophoresis on 1.8% agarose gel or (b) Western blot analyses for PARP-1 and ICAD. Closed arrowheads indicate the intact forms of PARP-1 (115 kDa) and ICAD (45 kDa). Open arrowheads show the bands of cleaved PARP-1 fragments (85 kDa)
Upregulation of DNaseγ upon BCR crosslinking in WEHI-231 cells
BCR ligation alters the expression levels of certain apoptosis-related molecules, and thus plays an important role in the control of BCR-mediated apoptosis in immature B cells.18, 19, 20 We therefore used the reverse transcription-polymerase chain reaction (RT-PCR) to investigate the changes in CAD and DNaseγ expression levels in response to BCR ligation in WEHI-231 cells. A time course study revealed that the expression of CAD was unaffected by BCR signaling. By contrast, the DNaseγ expression level increased shortly after BCR ligation, remained high for up to 6 h, and then decreased until 12 h, which coincided with the onset of apoptosis (Figure 6a). Quantitative real-time PCR revealed that DNaseγ expression had increased approximately sixfold at 3 h post-BCR ligation in both the control and ICAD-CR cells (Figure 6b). The activation of DNaseγ expression was also confirmed at the protein level by an activity gel assay (Figure 6a, bottom panel). Thus, BCR ligation was shown to induce the upregulation of DNaseγ before the onset of apoptosis.

BCR ligation upregulates DNaseγ expression in WEHI-231 cells. (a) Changes in DNaseγ and CAD expression levels in WEHI-231 cells induced by BCR ligation. WEHI-231 cells were cultured for the indicated times in the presence of 10 μg/ml anti-IgM antibody. The resulting cells were collected and subjected to RT-PCR (upper panel) and an activity gel assay (lower panel). cDNAs were normalized for β-actin expression. An arrowhead indicates the bands of the 33-kDa DNaseγ protein. (b) Quantitative real-time PCR analyses of the DNaseγ transcript in WEHI-231 cells. WEHI-231/Puro (Puro) and WEHI-231/ICAD-CR (ICAD-CR) cells were cultured for 3 h in the presence of anti-IgM antibody (10 μg/ml). The resulting cells were collected and subjected to real-time PCR. The expression levels of DNaseγ mRNA were normalized for GAPDH, and are indicated relative to the basal DNaseγ expression in unstimulated WEHI-231/Puro cells. All values are the mean±S.D. (n=3)
Inhibitor of DNaseγ suppresses nucleosomal ladder formation and nuclear fragmentation during BCR-mediated apoptosis in WEHI-231/ICAD-CR cells
We recently found that 4-(4,6-dichloro-[1, 3, 5]-triazin-2-ylamino)-2-(6-hydroxy-3-oxo-3H-xanthen-9-yl)-benzoic acid (DR396) strongly inhibited DNaseγ in vitro. Importantly, DR396 had little effect on DNase I, which is structurally related to DNaseγ, under the same assay conditions. Furthermore, DR396 selectively suppressed DNaseγ-mediated DNA-ladder formation, but not that induced by CAD, at the cellular level.21 We thus utilized DR396 to evaluate the role of DNaseγ in BCR-mediated apoptosis. Control (Puro) and ICAD-CR cells were stimulated with an anti-IgM antibody in the presence or absence of DR396. As shown in Figure 7a, DR396 strongly suppressed nucleosomal ladder formation without affecting the caspase-mediated cleavage of PARP-1, suggesting that it inhibited the functional apoptotic DNase without affecting the other apoptotic processes activated by BCR ligation. Although ladder formation was weakly detected in the control cells, DR396 almost completely suppressed the BCR-mediated DNA fragmentation in ICAD-CR cells. A control experiment showed that DR396 alone at doses up to 100 μM had no toxic effect on the cells (Figure 7a). Microscopic analysis revealed that DR396 also interfered with the nuclear fragmentation induced by BCR crosslinking in ICAD-CR cells (Figure 7b). Based on the specificity of DR396 and the fact that CAD was barely activated in ICAD-CR cells, we concluded that both nucleosomal ladder formation and nuclear fragmentation in BCR-mediated apoptosis were largely induced by DNaseγ in WEHI-231 cells.

Involvement of DNaseγ in BCR-mediated apoptosis in WEHI-231 cells. (a) Effects of DR396 on BCR ligation-induced apoptotic DNA fragmentation. WEHI-231/ICAD-CR (ICAD-CR) and WEHI-231/Puro (Puro) cells were treated with 10 μg/ml anti-IgM antibody for 24 h in the presence (+) or absence (−) of 100 μ M DR396. The resulting cells were collected and subjected to DNA electrophoresis on 1.8% agarose gel (upper panels), and Western blot analyses for the cleavage of ICAD (middle panels) and PARP-1 (lower panels). Closed arrowheads indicate the intact forms of PARP-1 (115 kDa) and ICAD (45 kDa). Open arrowheads show the bands of cleaved PARP-1 fragments (85 kDa). The bands representing the Myc and His-tagged ICAD-CR are marked by an asterisk. (b) Effects of DR396 on BCR ligation-induced nuclear morphological changes. WEHI-231/ICAD-CR cells (ICAD-CR) were treated with 10 μg/ml anti-IgM antibody for 24 h in the presence (+) or absence (−) of 100 μ M DR396. The resulting cells were stained with Hoechst 33342 and observed by DIC or fluorescence microscopy (Hoechst). The black bar represents 50 μm
Discussion
We previously investigated the roles of CAD and DNaseγ in cells that predominantly expressed either of these DNases;16, 17 however, it remained unclear how they contributed to apoptotic DNA fragmentation when they were expressed simultaneously. In the current study, we demonstrated that CAD and DNaseγ are both expressed in WEHI-231 cells, and are selectively utilized for nucleosomal DNA fragmentation depending on the cause of apoptosis.
Apoptotic DNA fragmentation induced by cytotoxic drugs was catalyzed by CAD. This apoptotic ladder formation was completely suppressed by the expression of ICAD-CR (Figure 2b). Apoptotic stimulation by cytotoxic drugs induces the activation of caspase-3 in various types of cell,22 and was shown here to promote ICAD cleavage in WEHI-231 cells (Figure 5b). Thus, these drugs appeared to induce apoptosis in conjunction with CAD activity. Unexpectedly, we found no evidence of the involvement of DNaseγ in chemically induced apoptosis in WEHI-231 cells. To date, several groups have independently reported that ectopically expressed DNaseγ is activated by cytotoxic drugs and induces DNA fragmentation in dying cells.14, 15, 23, 24 Furthermore, STS has been shown to produce DNA-ladder formation by activating endogenous DNaseγ in neuronally differentiated N1E-115 cells.17 The activation of DNaseγ expression occurs in certain cultured cell lines, such as C2C12, N1E-115, and PC12 cells, when they are exposed to differentiation conditions.16, 17 However, notably, the expression level of DNaseγ in unstimulated WEHI-231 cells was more than 10-times higher than the levels induced in the above-mentioned cells during differentiation (data not shown). Therefore, cells that constitutively express DNaseγ at high levels might have inherent protective mechanisms to keep its activity under strict control, and might require specific stimuli to allow it to function in apoptotic DNA fragmentation. This idea is supported by the observation that DNaseγ fails to catalyze DNA fragmentation in WEHI-231 cells that are degraded by necrotic stimuli (Figure 3).
We have shown that BCR-mediated apoptosis utilizes a unique DNA-fragmentation mechanism, which differs from that induced by cytotoxic drugs. DR396, but not ICAD-CR, effectively suppressed BCR-mediated apoptotic DNA fragmentation, indicating that the primary apoptotic DNase in this case is DNaseγ. The clonal deletion of self-reactive B cells occurs in immature B cells in which DNaseγ is highly expressed. This temporal correlation provides further evidence of DNaseγ involvement in BCR-mediated apoptosis.
Among the 14 caspases that occur in mammals, three (caspase-3, caspase-6, and caspase-7) act as executioners during the late stage of apoptosis.8 Caspase-7 is thought to be the most important caspase in BCR-mediated apoptosis (Figure 8a).25, 26 BCR ligation induces the selective activation of caspase-7 via two independent cascades: a mitchondria caspase-9 pathway and a calpain pathway.26, 27 Calpain, which is a Ca2+-dependent cysteine protease, is activated upon BCR ligation and catalyzes the processing of procaspase-7 into its active form.26 BCR ligation also triggers a sustained Ca2+ influx in WEHI-231 cells, which is thought to be an important trigger of calpain activation.28 Concomitant signaling through CD40 is known to rescue WEHI-231 cells from BCR-mediated apoptosis.29 Importantly, CD40 ligation induces the expression of the Bcl-2-like antiapoptotic protein A1 and the endogenous calpain inhibitor calpastatin.26, 30 A1 and calpastatin inhibit caspase-9 activation and calpain activity, respectively, thereby interfering with the activation of caspase-7.

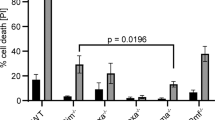
Caspase activation and its role in WEHI-231 apoptosis. (a) Activation of caspase-3 and caspase-7 during apoptosis induced by STS or BCR cross-linking. WEHI-231 cells that were untreated (Control), or treated with 0.1 μ M STS for 6 h or 10 μg/ml anti-IgM antibody for 24 h, were subjected to Western blot analysis for caspase-3 and caspase-7. The bands representing inactive precursors and activated forms are indicated by closed and open arrowheads, respectively. Asterisks show nonspecific bands that appeared on the blots. (b, c) Effects of caspase inhibitors on BCR-mediated apoptosis. WEHI-231 cells were treated with 10 μg/ml anti-IgM antibody for 24 h in the presence of the indicated doses of z-VAD-fmk or DEVD-fmk. The inhibitors were added to the culture 1 h before the addition of anti-IgM antibody. The resulting cells were collected and subjected to DNA electrophoresis on 1.8% agarose gel (b, upper panels), Western blot analysis for the cleavage of PARP-1 (b, lower panels), and a formazan production assay to evaluate the viability of the cells (c). The bands representing intact (115 kDa) and cleaved (85 kDa) PARP-1 are indicated by closed and open arrowheads, respectively. The viability of the cells is indicated relative to the result of a control experiment performed in the absence of anti-IgM antibody or caspase inhibitors. All values are the mean±S.D. (n=4)
Generally, caspase-3 is the primary executioner during mammalian cell apoptosis.22 However, there is growing evidence that BCR-mediated WEHI-231 apoptosis is not accompanied by caspase-3 activation (Figure 8a).25, 26, 31 This is supported by the fact that immature B cells from caspase-3-deficient mice undergo normal apoptosis.32 Both caspase-3 and caspase-7 catalyze the cleavage of PARP-1; however, the former cleaves ICAD more effectively in vitro, and endogenous caspase-7 barely promotes CAD activation in vivo.33, 34 These observations agree with our finding that PARP-1 cleavage is observed in both cytotoxic drug-mediated and BCR-mediated apoptosis, whereas the disappearance of intact ICAD occurs only in the former. Thus, WEHI-231 cells might primarily utilize DNaseγ during BCR-mediated apoptosis, because the cascade activated by BCR ligation is apparently less preferable for CAD activation. Elevated intracellular Ca2+ levels caused by BCR ligation might also affect the choice of apoptotic DNase, because this is an important cofactor for DNaseγ activity.14 We do not suggest that BCR-mediated DNA fragmentation is catalyzed exclusively by DNaseγ, because DNA-ladder formation is slightly impaired in ICAD-CR cells (Figure 4a) and might not be completely suppressed by DR396 in parental WEHI-231 cells (Figure 7a). Thus, trace amounts of CAD might also be activated during BCR-mediated apoptosis and partially contribute to DNA-ladder formation.
To clarify the role of caspase-7 in BCR-mediated DNaseγ activation, we tested the effects of the pan-caspase inhibitor z-VAD-fmk, and the specific inhibitor for caspase-3 and caspase-7 DEVD-fmk, on WEHI-231 apoptosis. z-VAD-fmk suppressed both PARP-1 cleavage and DNA fragmentation in a dose-dependent manner (Figure 8b). Moreover, z-VAD-fmk rescued the BCR-stimulated cells from death (Figure 8c). These results suggest that activation of a z-VAD-sensitive caspase is essential for the onset of BCR-mediated apoptosis. However, these data failed to reveal the importance of caspase-7 for BCR-mediated DNaseγ activation, because the absence of DNA fragmentation caused by z-VAD-fmk is probably a consequence of the inhibition of cell death. By contrast, DEVD-fmk inhibited PARP-1 cleavage without affecting the rate of BCR-mediated cell death (Figure 8c). DNA-ladder formation was slightly suppressed, provably owing to the inhibition of CAD activation, but remained similar to that observed in ICAD-CR cells (Figures 4a and 7a). Based on these results, together with the fact that STS-induced apoptosis does not activate DNaseγ (Figure 2b) regardless of the occurrence of caspase-7 activation (Figure 8a), we concluded that caspase-7 does not have a significant role in BCR ligation-induced DNaseγ activation.
Nuclear morphological changes have been used to define apoptotic cell death, and can be induced by acinus-dependent and DNase-dependent mechanisms.35, 36 In the latter, the nuclear changes occur in a sequential manner: chromosomal DNA initially accumulates around nuclear envelopes (stage I chromatin condensation) and is then fragmented into highly packed round masses (stage II chromatin condensation or nuclear fragmentation).36 CAD is indispensable for stage II chromatin condensation, as DT40 cells carrying a cad−/− mutation undergo stage I chromatin condensation but fail to proceed to stage II.37 Although it remains unclear whether CAD activity is required for stage I chromatin condensation, its essential role in nuclear morphological changes has been confirmed in cells prepared from ICAD-deficient mice.38 Our results obtained using cytotoxic drugs support these previous observations. However, we found that ICAD-CR failed to suppress stage II chromatin condensation during BCR-mediated apoptosis in WEHI-231 cells. We showed that BCR ligation-induced DNA-ladder formation was catalyzed by DNaseγ, such that DNA fragmentation was scarcely affected by ICAD-CR. Our results demonstrate the ability of DNaseγ to induce nuclear fragmentation or stage II chromatin condensation during apoptosis. Our data also indicate that the critical factor for stage II chromatin condensation is the occurrence of nucleosomal DNA fragmentation per se, rather than a specific type of apoptotic DNase being required for this process.
Our study has revealed that nucleosomal DNA fragmentation in WEHI-231 cells is catalyzed by CAD or DNaseγ depending on the cause of apoptosis. Apoptotic cells are engulfed by macrophages or neighboring cells, and are rapidly eliminated in vivo. If the genetic information of dying cells persists, it might transform phagocytes via horizontal transfer of the engulfed genes.39 Furthermore, defects in DNA waste clearance can cause serious pathological conditions (such as autoimmune diseases, lethal anemia, and impaired thymic development) by disturbing the immune system.7 Therefore, preparing for possible death by installing multiple apoptotic DNases could be an excellent cellular strategy to avoid such hazardous situations.
Materials and Methods
Cell culture
Murine B cell lines (18–81 (pre-B), WEHI-231 (immature B), CH31 (immature B), WEHI-279 (mature B), A20 (mature B), and J558L (plasma)) were cultured in RPMI-1640 supplemented with 10% fetal calf serum, 50 μM of 2-mercaptoethanol, 100 U/ml penicillin, and 100 μg/ml streptomycin.
Preparation of spleen B cells
C57BL/6 mice (18 weeks old) were irradiated with X-rays at 5 Gy to remove all lymphocytes. After 13 days of irradiation, the spleens in which newly generated IgMhigh IgDlow B cells had accumulated were removed from the irradiated and age-matched control animals. The spleen cells were suspended in FACS buffer (Dulbecco's modified phosphate-buffered saline containing 0.1% bovine serum albumin, and 0.1% sodium azide), passed through a 70-μm pore size nylon filter (Falcon), and washed twice with the same buffer. After removal of the red blood cells by suspension in a hypotonic buffer (0.15 M NH4Cl, 1 mM KHCO3, and 0.1 mM EDTA (pH 7.3)), they were further treated with an anti-Thy1.1 antibody (Pharmingen) in the presence of a Low-Tox-M rabbit complement (Cedarlane), and the T cells were removed. The resulting cells were washed twice, resuspended in FACS buffer and subjected to flow cytometry or an activity gel assay.
Cell staining and flow cytometry
The spleen B cells were analyzed by flow cytometry to examine the expression profiles of the indicated cell-surface antigens. Single-cell suspensions, prepared as described above, were incubated with antibodies specific for the indicated antigens for 30 min on ice. The antibodies used were as follows: PE-anti B220 (1 : 1000 dilution; Pharmingen); FITC-anti-CD3 (1 : 1000 dilution; Pharmingen); PE-anti-IgM (1 : 1000 dilution; Southern Biotechnology); and FITC-anti-IgD (1 : 1000 dilution; Southern Biotechnology). The resulting cells were washed twice with the same buffer and analyzed by FACScan using Cellquest software (Becton Dickinson).
Gene-expression analyses
Northern blot analysis was performed as described previously, with some modifications.13 Briefly, 15 μg total RNA was separated by 1% agarose-formamide gel electrophoresis and transferred to a Hybond-N+ membrane (Amersham). The blot was hybridized with a 32P-labeled cDNA fragment for the entire open reading frame of mouse DNaseγ (GenBank U76110) using PerfectHyb hybridization solution (Toyobo), and was visualized using a BAS1500 imaging system (Fuji Film). The resulting blot was boiled in a stripping solution (sterile H2O containing 0.5% SDS) for 5 min, and was repeatedly hybridized for ICAD (GenBank AB009375) and CAD (GenBank AB009377). RT-PCR analysis was performed as described previously.16 PCR amplification was carried out for the indicated number of cycles, each of which consisted of denaturation at 94°C for 30 s, annealing at 62°C for 30 s, and extension at 72°C for 1 min. The primers used were as follows: CAD (34 cycles), 5′-CAGGACCCATCCTGGTTTGAAGGT-3′ (sense) and 5′-GATCCTGTCTGAGGCCGATAGATC-3′ (antisense); ICAD (30 cycles), 5′-GGCCATTGATAAGTCCCTGAC-3′ (sense) and 5′-CCTGAGTGAATGCAAGCTCTG-3′ (antisense); DNaseγ (32 cycles), 5′-CACGTACAAAGAGCAGTATGC-3′ (sense) and 5′-CGAATGTTCTGCCAGGCCTTC-3′ (antisense); and β-actin (25 cycles), 5′-CACCTTCTACAATGAGCTGCG-3′ (sense) and 5′-CTTGCTGATCCACATCTGCTG-3′ (antisense). To quantify the DNaseγ transcripts, real-time PCR analysis was performed using a QuantiTect SYBR Green PCR kit (Qiagen) in an ABI Prism 7700 sequence detector (Applied Biosystems). All assays were performed in triplicate and normalized for β-actin mRNA.
Assay for DNaseγ activity
DNaseγ activity was analyzed by an activity gel assay as described previously.16 The in-gel reaction was performed in a reaction buffer (10 mM Tris-HCl (pH 7.8), 1 mM 2-mercaptoethanol, 3 mM CaCl2, and 3 mM MgCl2) overnight at 37°C. After observing the DNase activity, the resulting gel was subjected to Coomassie Brilliant Blue staining to confirm equal lane loadings (data not shown).
Western blot analysis
Western blot analysis was performed using rabbit polyclonal antibodies for PARP-1 (1 : 1000 dilution; Wako), ICAD (1 : 1000 dilution; Medical and Biological Laboratories), caspase-3 (1 : 1000 dilution; Cell Signaling), caspase-7 (1 : 1000 dilution; Cell Signaling), and β-actin (1 : 40 000 dilution; Sigma) as described previously.16, 17
Establishment of WEHI-231/ICAD-CR cells
Exponentially growing WEHI-231 cells were washed twice with PBS and resuspended in OptiMEM (Gibco) supplemented with 20% FCS at a concentration of 5 × 106/ml. Aliquots of the cell suspension (500 μl) were chilled on ice for 10 min. Then, 20 μg of the expression vector pCAG/ICAD-CR-Myc-His17 was added to each aliquot, which was transfected by electroporation at 250 V and 800 μF using a Cell-Porator electroporation device (Gibco). After incubation on ice for 10 min, the resulting cells were diluted with 5 ml culture medium and cultured overnight. Cells that were stably integrated by the transgene were selected based on puromycin resistance (250 ng/ml). The resulting cells were cloned by two cycles of colony isolation, and several independent clones were obtained. Subcultures of the clones were subjected to Western blot analysis, and cells expressing high levels of the ICAD-CR protein were selected. As a consequence, we obtained three clones of WEHI-231/ICAD-CR cells: clones 1.10, 1.17, and 3.02. We observed no difference in apoptotic phenotypes among the clones (data not shown); the results obtained with clone 1.10 are shown in the figures. A mock transfectant, WEHI-231/puro cells, was obtained by the same method (with the exception that selection was by ICAD-CR expression) and used in the control experiments.
Apoptosis assays
WEHI-231 cells (1 × 105/ml) were treated with various apoptosis inducers for the indicated times. All of the chemical apoptosis inducers were purchased from Sigma. The caspase inhibitors, z-VAD-fmk and DEVD-fmk, were purchased from Chemicon. The AffiniPure F(ab′)2 fragment goat anti-mouse IgM antibody was obtained from Jackson Immuno Research. The resulting cells were collected and subjected to the apoptosis assays. DNA-ladder formation was analyzed by 1.8% agarose gel electrophoresis as described previously.16 Cellular and nuclear morphological changes were observed under a fluorescence microscope (Olympus, BX60). Nuclear staining with Hoechst 33342 dye (Molecular probes) was performed as described previously.16 The cell viability was evaluated using the CellTiter 96 AQueous One Solution Cell Proliferation Assay kit (Promega) according to the manufacturer's protocol. This assay quantifies the ability of a cell to convert a tetrazolium compound into a colored formazan product by measuring the absorbance at 490 nm with a 96-well plate reader.
Abbreviations
- BCR:
-
B-cell receptor
- BSA:
-
bovine serum albumin
- CAD:
-
caspase-activated DNase
- cDNA:
-
complementary DNA
- CHX:
-
cycloheximide
- DFF:
-
DNA-fragmentation factor
- DIC:
-
differential interference contrast
- DPBS:
-
Dulbecco's modified phosphate-buffered saline
- DR396:
-
4-(4,6-dichloro-[1, 3, 5]-triazin-2-ylamino)-2-(6-hydroxy-3-oxo-3H-xanthen-9-yl)-benzoic acid
- Etp:
-
etoposide
- ICAD:
-
inhibitor of CAD
- Ig:
-
immunoglobulin
- mRNA:
-
messenger RNA
- PARP-1:
-
poly (ADP-ribose) polymerase-1
- rRNA:
-
ribosomal RNA
- RT-PCR:
-
reverse transcription-polymerase chain reaction
- S.D.:
-
standard deviation
- STS:
-
staurosporine
- Tg:
-
thapsigargin
- Tm:
-
tunicamycin
References
Nemazee D, Hogquist KA . Antigen receptor selection by editing or downregulation of V(D)J recombination. Curr Opin Immunol 2003; 15: 182–189.
Tsubata T, Honjo T . B cell tolerance and autoimmunity. Rev Immunogenet 2000; 2: 18–25.
Hasbold J, Klaus GG . Anti-immunoglobulin antibodies induce apoptosis in immature B cell lymphomas. Eur J Immunol 1990; 20: 1685–1690.
Benhamou LE, Cazenave PA, Sarthou P . Anti-immunoglobulins induce death by apoptosis in WEHI-231 B lymphoma cells. Eur J Immunol 1990; 20: 1405–1407.
Meier P, Finch A, Evan G . Apoptosis in development. Nature 2000; 407: 796–801.
Ziegler U, Groscurth P . Morphological features of cell death. News Physiol Sci 2004; 19: 124–128.
Samejima K, Earnshaw WC . Trashing the genome: the role of nucleases during apoptosis. Nat Rev Mol Cell Biol 2005; 6: 677–868.
Fuentes-Prior P, Salvesen GS . The protein structures that shape caspase activity, specificity, activation and inhibition. Biochem J 2004; 384: 201–232.
Broker LE, Kruyt FA, Giaccone G . Cell death independent of caspases: a review. Clin Cancer Res 2005; 11: 3155–3162.
Liu X, Zou H, Slaughter C, Wang X . DFF, a heterodimeric protein that functions downstream of caspase-3 to trigger DNA fragmentation during apoptosis. Cell 1997; 89: 175–184.
Enari M, Sakahira H, Yokoyama H, Okawa K, Iwamatsu A, Nagata S . A caspase-activated DNase that degrades DNA during apoptosis, and its inhibitor ICAD. Nature 1998; 391: 43–50.
Shiokawa D, Ohyama H, Yamada T, Takahashi K, Tanuma S . Identification of an endonuclease responsible for apoptosis in rat thymocytes. Eur J Biochem 1994; 226: 23–30.
Shiokawa D, Tanuma S . Molecular cloning and expression of a cDNA encoding an apoptotic endonuclease DNaseγ. Biochem J 1998; 332: 713–720.
Shiokawa D, Tanuma S . Characterization of human DNase I family endonucleases and activation of DNaseγ during apoptosis. Biochemistry 2001; 40: 143–152.
Yakovlev AG, Wang G, Stoica BA, Simbulan-Rosenthal CM, Yoshihara K, Smulson ME . Role of DNAS1L3 in Ca2+- and Mg2+-dependent cleavage of DNA into oligonucleosomal and high molecular mass fragments. Nucleic Acids Res 1999; 27: 1999–2005.
Shiokawa D, Kobayashi T, Tanuma S . Involvement of DNaseγ in apoptosis associated with myogenic differentiation of C2C12 cells. J Biol Chem 2002; 277: 31031–31037.
Shiokawa D, Tanuma S . Differential DNases are selectively used in neuronal apoptosis depending on the differentiation state. Cell Death Differ 2004; 11: 1112–1120.
Fischer G, Kent SC, Joseph L, Green DR, Scott DW . Lymphoma models for B cell activation and tolerance. X. Anti-mu-mediated growth arrest and apoptosis of murine B cell lymphomas is prevented by the stabilization of myc. J Exp Med 1994; 179: 221–228.
Merino R, Grillot DA, Simonian PL, Muthukkumar S, Fanslow WC, Bondada S et al. Modulation of anti-IgM-induced B cell apoptosis by Bcl-xL and CD40 in WEHI-231 cells. Dissociation from cell cycle arrest and dependence on the avidity of the antibody-IgM receptor interaction. J Immunol 1995; 155: 3830–3838.
Wu M, Bellas RE, Shen J, Sonenshein GE . Roles of the tumor suppressor p53 and the cyclin-dependent kinase inhibitor p21WAF1/CIP1 in receptor-mediated apoptosis of WEHI 231 B lymphoma cells. J Exp Med 1998; 187: 1671–1679.
Sunaga S, Kobayashi T, Yoshimori A, Shiokawa D, Tanuma S . A novel inhibitor that protects apoptotic DNA fragmentation catalyzed by DNase gamma. Biochem Biophys Res Commun 2004; 325: 1292–1297.
Porter AG, Janicke RU . Emerging roles of caspase-3 in apoptosis. Cell Death Differ 1999; 6: 99–104.
Boulares AH, Ren T . Mechanism of acetaminophen-induced apoptosis in cultured cells: roles of caspase-3, DNA fragmentation factor, and the Ca2+ and Mg2+ endonuclease DNAS1L3. Basic Clin Pharmacol Toxicol 2004; 94: 19–29.
Boulares H, Zoltoski A, Kandan S, Akbulut T, Yakovlev A, Oumouna M . Correlation between decreased sensitivity of the Daudi lymphoma cells to VP-16-induced apoptosis and deficiency in DNAS1L3 expression. Biochem Biophys Res Commun 2006; 341: 653–662.
Bras A, Ruiz-Vela A, Gonzalez de Buitrago G, Martinez-A C . Caspase activation by BCR cross-linking in immature B cells: differential effects on growth arrest and apoptosis. FASEB J 1999; 13: 931–944.
Ruiz-Vela A, Gonzalez de Buitrago G, Martinez-A C . Implication of calpain in caspase activation during B cell clonal deletion. EMBO J 1999; 18: 4988–4998.
Herold MJ, Kuss AW, Kraus C, Berberich I . Mitochondria-dependent caspase-9 activation is necessary for antigen receptor-mediated effector caspase activation and apoptosis in WEHI 231 lymphoma cells. J Immunol 2002; 168: 3902–3909.
Nam JH, Yoon SS, Kim TJ, Uhm DY, Kim SJ . Slow and persistent increase of [Ca2+]c in response to ligation of surface IgM in WEHI-231 cells. FEBS Lett 2003; 535: 113–118.
Tsubata T, Wu J, Honjo T . B-cell apoptosis induced by antigen receptor crosslinking is blocked by a T-cell signal through CD40. Nature 1993; 364: 645–648.
Kuss AW, Knodel M, Berberich-Siebelt F, Lindemann D, Schimpl A, Berberich I . A1 expression is stimulated by CD40 in B cells and rescues WEHI 231 cells from anti-IgM-induced cell death. Eur J Immunol 1999; 29: 3077–3088.
Mlinaric-Rascan I, Turk B . B cell receptor-mediated nuclear fragmentation proceeds in WEHI 231 cells in the absence of detectable DEVDase and FRase activity. FEBS Lett 2003; 553: 51–55.
Woo M, Hakem R, Soengas MS, Duncan GS, Shahinian A, Kagi D et al. Essential contribution of caspase 3/CPP32 to apoptosis and its associated nuclear changes. Genes Dev 1998; 12: 806–819.
Wolf BB, Schuler M, Echeverri F, Green DR . Caspase-3 is the primary activator of apoptotic DNA fragmentation via DNA fragmentation factor-45/inhibitor of caspase-activated DNase inactivation. J. Biol Chem 1999; 274: 30651–30656.
Slee EA, Adrain C, Martin SJ . Executioner caspase-3, -6, and -7 perform distinct, non-redundant roles during the demolition phase of apoptosis. J Biol Chem 2001; 276: 7320–7326.
Sahara S, Aoto M, Eguchi Y, Imamoto N, Yoneda Y, Tsujimoto Y . Acinus is a caspase-3-activated protein required for apoptotic chromatin condensation. Nature 1999; 401: 168–173.
Samejima K, Tone S, Kottke TJ, Enari M, Sakahira H, Cooke CA et al. Transition from caspase-dependent to caspase-independent mechanisms at the onset of apoptotic execution. J Cell Biol 1998; 143: 225–239.
Samejima K, Tone S, Earnshaw WC . CAD/DFF40 nuclease is dispensable for high molecular weight DNA cleavage and stage I chromatin condensation in apoptosis. J Biol Chem 2001; 276: 45427–45432.
Zhang J, Liu X, Scherer DC, van Kaer L, Wang X, Xu M . Resistance to DNA fragmentation and chromatin condensation in mice lacking the DNA fragmentation factor 45. Proc Natl Acad Sci USA 1998; 95: 12480–12485.
Holmgren L, Szeles A, Rajnavolgyi E, Folkman J, Klein G, Ernberg I et al. Horizontal transfer of DNA by the uptake of apoptotic bodies. Blood 1999; 93: 3956–3963.
Acknowledgements
This work was funded, in part, by a Grant-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology of Japan.
Author information
Authors and Affiliations
Corresponding author
Additional information
Edited by JA Trapani
Rights and permissions
About this article
Cite this article
Shiokawa, D., Shika, Y., Araki, S. et al. Stage-specific expression of DNaseγ during B-cell development and its role in B-cell receptor-mediated apoptosis in WEHI-231 cells. Cell Death Differ 14, 992–1000 (2007). https://doi.org/10.1038/sj.cdd.4402086
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.cdd.4402086
