
Abstract
Apoptosis is a distinct form of cell death, which requires energy. Here, we made real-time continuous measurements of the cytosolic ATP level throughout the apoptotic process in intact HeLa, PC12 and U937 cells transfected with the firefly luciferase gene. Apoptotic stimuli (staurosporine (STS), tumor necrosis factor α (TNFα), etoposide) induced significant elevation of the cytosolic ATP level. The cytosolic ATP level remained at a higher level than in the control for up to 6 h during which activation of caspase-3 and internucleosomal DNA fragmentation took place. When the STS-induced ATP response was abolished by glucose deprivation-induced inhibition of glycolysis, both caspase activation and DNA laddering were completely inhibited. Annexin V-binding induced by STS or TNFα was largely suppressed by glycolysis inhibition. Thus, it is suggested that the cells die with increased cytosolic ATP, and elevation of cytosolic ATP level is a requisite to the apoptotic cell death process.
Similar content being viewed by others
Introduction
Apoptosis is a genetically programmed cell death process, which is triggered by external and internal signals. This distinct form of cell death plays an essential role in tissue development and homeostasis,1 and also in the elimination of cancerous, virally infected or degenerated cells.2 At present, there is no question that apoptosis requires energy, because it is a highly regulated process involving a number of ATP-dependent steps such as caspase activation, enzymatic hydrolysis of macromolecules, chromatin condensation, bleb formation and apoptotic body formation.3, 4, 5, 6, 7, 8, 9 Depletion of cellular ATP was found to cause switching of the form of cell death, from apoptotic cell death triggered by a variety of stimuli to necrotic cell death.5, 10, 11, 12, 13, 14 Thus, it is now well accepted that the level of intracellular ATP determines whether the cell dies by apoptosis or necrosis.4, 7
The total ATP level in different cell types undergoing apoptosis has been measured by a luciferin–luciferase assay following lysis of the cells.5, 12, 13, 15, 16, 17, 18, 19 Although most studies demonstrated that apoptotic stimulation reduced, to a certain extent, the total cellular ATP level,13, 15, 17, 18, 19 several studies found that levels of total ATP5, 16, 20 were unchanged during early stages of apoptosis. Such variability in measured ATP levels might be, at least in part, due to differences in the time period of apoptotic stimulation and/or the degree of contamination by cells that have already died and lost ATP. It should also be noted that ATP may not be uniformly distributed throughout the cell body.21, 22 Thus, it is likely that the total ATP levels hitherto measured do not accurately represent the cytosolic ATP levels.
In the present study, we made continuous measurements of the cytosolic ATP level in intact cells throughout the apoptotic process, up to their death. Our results show that cytosolic ATP within cells undergoing apoptosis is maintained at a level higher than in control cells even as caspase activation and DNA laddering are occurring, during the final stages of cell death. Also, we suggest that elevation of the cytosolic ATP level is a prerequisite to the apoptotic cell death process.
Results
Elevation of the cytosolic ATP level was induced by apoptotic stimulation
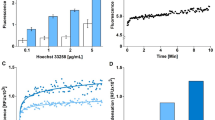
HeLa cells have been shown to respond to staurosporine (STS), a drug that induces mitochondrion-mediated apoptosis, with an apoptotic volume decrease (AVD), caspase-3 activation and cell death, which start 0–1, 2–3 and 3–4 h, respectively, after treatment.23 Here, we measured the cytosolic ATP level of luciferase-expressing HeLa cells incubated in Ringer solution, during the proapoptotic stage and up to 70 min after STS application. As shown in Figure 1a, STS (4 μM) promptly caused an increase in cytosolic ATP-dependent luminescence. The signal reached a maximum value (227±16%, n=9: P=3.1 × 10−5) within 15–30 min and then gradually decreased to a level of around 150–200%. In contrast, addition of an equal amount of dimethyl sulfoxide (DMSO), the vehicle for STS, did not alone cause any change in the luminescence signal (data not shown, n=7).

Cytosolic ATP rise induced by STS, etoposide and TNFα in luciferase-expressing HeLa, PC12 and U937 cells incubated in Ringer solution. (a–c) Representative time course of luminescence signals from HeLa cells before and during continuous exposure to membrane-permeable luciferin (100 μM) and STS (4 μM) in control conditions (a) and in the presences of 50 μM zVAD-fmk (b) or 5 mM Ba2+ (c). (d) Concentration–response curve of the STS effect on the luminescence signal. The peak level of luminescence was normalized to the level before addition of STS. Mean values±S.E.M. of triplicate measurements are plotted. The solid line is a fit to a Hill equation with EC50=0.46±0.09, a Hill coefficient of 1.8 and Vmax=223±5.5%. (e–h) Representative time course of luminescence signals from PC12 cells (e) and from U937 cells (f) before and during continuous exposure to 4 μM STS, as well as from HeLa cells before and during continuous exposure to 100 μM etoposide (g) and of 20 ng/ml TNFα+20 μg/ml CHX (h) in the presence of luciferin (100 μM). The absolute intensity of signals varied with the level of luciferase expression
The STS-induced rise of cytosolic ATP signal was not affected by a pancaspase inhibitor, zVAD-fmk (50 μM added at 10 min before and during application of STS), as shown in Figure 1b. The peak value of relative luminescence in the presence of zVAD-fmk (253±20%, n=7) was not significantly different from that in the absence of zVAD-fmk but presence of the vehicle (DMSO) alone (260±16%, n=7: P=0.7767). This fact indicates that the STS-induced ATP response is an upstream event to caspase activation.
An essentially similar ATP signal response (255±12%, n=5) was induced by STS in the presence of a K+ channel blocker, Ba2+ (5 mM, Figure 1c), which was shown to block STS-induced AVD in a variety of cell types including HeLa cells.23 Thus, it appears that the cytosolic ATP rise is not the secondary effect of AVD.
The peak ATP signal response to STS was concentration-dependent with an EC50 of 0.46±0.09 μM and a Hill coefficient of 1.81±0.58, as summarized in Figure 1d. The basal concentration of cytosolic ATP under control conditions was estimated to be 1.25±0.15 mM by calibration procedures. This value is close to the values observed for MIN6 cells transfected with luciferase24 and for COSm6 cells transfected with a mutant KATP channel.25 The peak ATP concentration of STS-induced response was over several millimoles, although this could not be evaluated accurately, because the signal came close to the saturation level in the calibration curve.
To examine whether STS induces an increase in the total ATP of nontransfected HeLa cells, we used a conventional luciferin–luciferase assay to measure the total ATP content in cell lysates. A sizeable increase in the total ATP content was observed after 30-min treatment with 4 μM STS, and the mean value (6.54±0.28 fmol/cell, n=12) was significantly greater (P=0.0020) than measured in control experiments using DMSO (4.91±0.37 fmol/cell, n=11). The fact that the percent increase in total ATP (133%) was less than the percent increase in the cytosolic ATP signal (227%) may suggest that the STS-induced ATP increase in the cytosol was more marked than in noncytosolic compartments, and/or that the total ATP level was underestimated by some unknown factors (such as transient rise in intracellular calcium levels during cell lysis) involved in the assay procedures.
Next, we examined whether the STS-induced cytosolic ATP rise is specific to HeLa cells. We employed rat pheochromocytoma PC12 and human lymphoid U937 cells, because these cell lines have been shown to respond to STS with a series of apoptotic events with time courses similar to that for HeLa cells.23 For these cells transiently expressing luciferase, a similar STS-induced increase in the luminescence signal could be detected (Figures 1e and f). The STS-induced ATP signal response reached the peak value of 230±2% (n=5: P=3.3 × 10−7) in PC12 cells and 190±13% (n=7: P=3.7 × 10−4) in U937 cells. These results indicate that the STS-induced cytosolic ATP rise is not cell type-specific.
We then examined whether the cytosolic ATP response is specific to STS. An inducer of DNA damage-mediated apoptosis, etoposide (100 μM), was also found to increase the cytosolic ATP signal in HeLa cells (Figure 1g) with a time course similar to that for STS (Figure 1a). The peak value was 171±5% (n=5: P=1.33 × 10−4), which corresponds to the ATP concentration of 5.0±0.7 mM. A rise in cytosolic ATP also occurred when an inducer of death receptor-mediated apoptosis, tumor necrosis factor α (TNFα) (20 ng/ml), was administered to HeLa cells in combination with cycloheximide (CHX, 20 μg/ml) (Figure 1(h). Although an increase in the signal intensity was consistently observed in all experiments, the response had a lower peak value (126±5%, n=6: P=0.0041; corresponding to 2.0±0.1 mM ATP) and a slower time course than for STS (Figure 1a) or etoposide (Figure 1g). On the whole, however, these results indicate that the cytosolic ATP rise is a phenomenon common to apoptosis induced by mitochondrion-, DNA damage- and death receptor-mediated mechanisms.
Elevation of cytosolic ATP levels continued even after activation of caspase-3 and DNA laddering
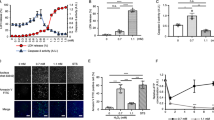
To examine how long the STS-induced ATP elevation persists, we observed the time course of changes in the cytosolic ATP-dependent luminescence in HeLa cells, which were incubated for varying lengths of time in culture medium (not Ringer solution) containing 4 μM STS. As shown in Figure 2a (squares), STS-induced elevation of cytosolic ATP signal was found to continue for up to 4 h and then to gradually decrease, reaching the zero level at about 10 h. A similar time course of elevation of cytosolic ATP signal was observed when the cells were incubated in culture medium free of STS for 1–10 h after a 25-min pretreatment with 4 μM STS, as shown in Figure 2a (circles). Marked elevation of the cytosolic ATP signal was found even just after washout of STS, and was maintained for up to 4 h. The ATP signal then gradually declined, falling below the control level after 7 h and down to a level slightly above the background level at 10 h. It must be noted that the ATP signal of the STS-exposed cell population returned to the control level at approximately 6 h when cell viability was only about 50% (see below). In any case, these experiments demonstrated that the cytosolic ATP level in both STS-treated and STS-pretreated cells is higher than in control cells for up to 6 h.

Comparison of time courses of the cytosolic ATP level and apoptotic events. Cytosolic ATP-dependent luminescence signals (a), cell death (b), caspase-3 activation (c) and DNA laddering (d) were monitored in luciferase-expressing HeLa cells incubated in culture medium after a 25-min pretreatment with 4 μM STS (filled circles), or under continuous stimulation by 4 μM STS (filled squares) in (a) after the same pretreatment. Open symbols represent control data (c) that were collected before the 25-min pretreatment and are plotted at t=−0.4. Each symbol represents the mean value (±S.E.M.) of five (a), 16 (b) or three (c) experiments. The data shown in (a) and (b) were obtained from independent experiments. The data shown in (d) represent triplicate experiments, the samples of which were the same as those in (c). M: 100-bp markers
As shown in Figure 2b, loss of viability of luciferase-expressing HeLa cells after a 25-min pretreatment with STS was found to proceed with a time course similar to that of the fall in cytosolic ATP signal (Figure 2a). This suggests that the gradual decrease in cytosolic ATP was associated with an increase in the number of dead cells. As shown in Figure 2c and d, activation of caspase-3 and internucleosomal DNA fragmentation started within 3–4 h after STS pretreatment. These results indicate that execution events in apoptosis, such as caspase activation and DNA laddering, take place in the presence of excess cytosolic ATP, and that this highly energized cell state is not restricted to the early proapoptotic stage but extends through the executive apoptosis stage.
When an inducer of death receptor-mediated apoptosis, TNFα (20 ng/ml), was applied to HeLa cells in combination with CHX (20 μg/ml) in the culture medium (not in Ringer solution as in Figure 1h), the cytosolic ATP signal increased above the control level and remained high up to 5 h (Figure 3a). The following decline in ATP-dependent luminescence paralleled the change in cell viability (Figure 3b). It is noteworthy that after 8 h of TNFα/CHX treatment, measured ATP signal remained at approx. 80% of the initial level, while about 40% of cells have already died, suggesting that remaining cells had high cytosolic ATP concentrations. Caspase-3 activation and DNA laddering in these experimental conditions were observed already at 4 h in our previous study.23

Effect of TNFα on the cytosolic ATP level and cell death in luciferase-expressing HeLa cells. Cytosolic ATP-dependent luminescence signals (a) and cell viability (b) were monitored in luciferase-expressing HeLa cells incubated in culture medium in the presence of 20 ng/ml TNFα+20 μg/ml CHX. Open symbols represent data from controls (c) before adding TNFα+CHX. Each symbol represents the mean value (±S.E.M.: vertical bar) of five (a) and five to eight (b) experiments
STS-induced rise in cytosolic ATP depends on the glycolytic reaction
When HeLa cells were incubated in normal Ringer solution containing glucose and supplemented with a mitochondrial Fo, F1-ATPase inhibitor, oligomycin (10 μM), abolishment of the STS-induced rise in ATP signal was never observed, although the peak response became less prominent and less prolonged (Figure 4a) compared to under control conditions (Figure 1a). The mean peak response was 151±7% (n=7: P=0.0010) of the control basal level. When the glycolytic reaction was blocked using 2-deoxy-D-glucose (2DG, 20 mM) in glucose-free Ringer solution, the STS-induced ATP response was abolished (Figure 4b). The peak level was 104±5% (n=12: P=0.408) of the control basal level, and after reaching the peak, the ATP signal was maintained near normal levels. When both respiratory and glycolytic reactions were inhibited using 2DG and oligomycin under glucose-free conditions, STS brought about a rapid decrease in the ATP signal (Figure 4c) to 58±4% (n=7: P=1.5 × 10−4). When glucose-free solution containing 2DG and supplemented with a substrate of mitochondrial oxidative phosphorylation, pyruvate (5 mM) was used, the STS-induced ATP signal response was partially restored (to 119±6%, n=12: P=0.010), as shown in Figure 4d. These results indicate that STS-induced elevation of the cytosolic ATP level is supported mainly by glycolysis in the cytosol and partly by oxidative phosphorylation in the mitochondria.

Effects of metabolic inhibition on the STS-induced ATP response in luciferase-expressing HeLa cells. Representative time courses of luminescence signals emitted before and during continuous exposure to luciferin (100 μM) and STS (4 μM), for HeLa cells incubated in: (a) normal Ringer solution containing glucose and supplemented with 10 μM oligomycin, (b) glucose-free Ringer solution supplemented with 10 mM 2DG and (c, d) glucose-free Ringer solution supplemented with 20 mM 2DG and 10 μM oligomycin or 5mM pyruvate
Elevated level of cytosolic ATP may be a prerequisite to apoptotic cell death
To answer the question as to whether elevation of the cytosolic ATP level is a prerequisite to apoptotic cell death, we observed caspase-3 activation, DNA laddering and Annexin V binding after 25-min pretreatment of HeLa cells with STS under conditions in which glycolysis and/or respiratory ATP generation were inhibited by applying 2DG in the absence of glucose and/or by applying oligomycin only for 25 min during the STS treatment. When STS-induced ATP rise was prevented by inhibition of glycolysis only, caspase-3 activation (Figure 5a: diamonds) and DNA fragmentation (Figure 5b: diamond) were abolished, and Annexin V binding was also significantly diminished (Figure 5c: STS+2DG), even though the cytosolic ATP signal level was maintained near normal after STS application (Figure 4b). When the STS-induced ATP rise was partially suppressed by inhibition of respiratory reactions with oligomycin, STS-induced activation of caspase-3 was significantly delayed and DNA laddering was partially inhibited, as shown in Figures 5a and b (triangles). This result is in good agreement with a previous observation in Jurkat cells treated with STS and oligomycin.26 However, Annexin V binding measured after 1 h was little affected by oligomycin alone (Figure 5c: STS+oligomycin). When STS-induced ATP responses were abolished and cytosolic ATP signal was reduced below the normal level by inhibiting both glycolysis (using 2DG in glucose-free solution) and respiration (with oligomycin), no biochemical signs of executive apoptosis events were detected by assays for caspase-3 activation (Figure 5a: squares) and DNA fragmentation (Figure 5b: square). Also, Annexin V binding was significantly reduced by metabolic inhibition (Figure 5c: STS+2DG+oligomycin). Annexin V binding was increased, in a zVAD-fmk-dependent manner, by treatment not only with STS but also with TNFα/CHX (Figures 5c and d). Similar suppression of TNFα-induced Annexin V-binding was observed not only by application of 2DG or 2DG+oligomycin but also by oligomycin alone for 1 h during the treatment with TNFα+CHX (Figure 5d). The population of propidium iodide (PI)-positive necrotic cells, which was evaluated 1 h after the cells were transferred to the normal glucose-containing Ringer solution, was slightly increased by treatment with STS or TNFα+CHX (to approx. 6%: Figure 5e), but this was not significantly affected by cotreatment (for 25 min only) with oligomycin, 2DG in glucose-free solution or 2DG+oligomycin in glucose-free solution (Figure 5e). Based on these results, it is suggested that elevation of the cytosolic ATP level is an absolute requirement for the events in apoptotic cell death.

Effects of metabolic inhibitors on STS-induced activation of caspase-3 (a), STS-induced DNA laddering (b), STS-induced Annexin V binding (c), TNFα-induced Annexin V binding (d) and STS- and TNFα-induced PI staining (e) in luciferase-expressing HeLa cells. HeLa cells were pretreated with 4 μM STS for 25 min or with 20 ng/ml TNFα (+20 μg/ml CHX) for 60 min in the absence or presence of 10 μM oligomycin and/or 10 mM 2DG, and then transferred to the normal Ringer solution or to the culture medium, respectively. zVAD-fmk (50 μM) was added to the solutions 15 min prior to application of apoptotic inducers and throughout the experiments. The cells were then incubated for 2–6 h for caspase-3 activity (a), for 6 h for DNA laddering (b) and for 1 h for assays of Annexin V binding (c and d) and of PI exclusion (e). Data in (a) are the mean values (±S.E.M.) of six experiments, and data in (b) are representative of the same number of experiments. In (a) and (b), open and filled circles represent negative control data (STS not applied) and positive control data (STS applied), respectively, and triangles, diamonds and squares represent data with STS+oligomycin, STS+2DG and STS+oligmycin+2DG, respectively. M: 100-bp markers
Discussion
The firefly luciferin/luciferase reaction has been of great utility in monitoring ATP concentrations as measured by released photon count, because luciferase catalyzes the conversion of ATP, luciferin and O2 to AMP, oxyluciferin and CO2 with simultaneous generation of a photon.27 Luciferase introduced inside the cells has been successfully used to continuously monitor the intracellular ATP concentration and its changes in various physiological conditions.24, 28 In the present study, we used this powerful technique to measure time-dependent changes in the cytosolic ATP level after apoptotic stimulation in three different cell types transfected with the firefly luciferase gene. For cells incubated in Ringer solution, cytosolic ATP was found to increase (to over several millimoles) upon application of an apoptotic inducer (STS, etoposide or TNFα), reaching a peak level within around 30 min, and slowly decreasing thereafter. At the STS concentration of 100 nM (conventionally used as a PKC inhibitor), ATP-increasing effect was not observed. The cytosolic ATP increased significantly at the STS concentrations of 0.5 μM and more. Since micromolar dose of STS was necessary to induce apoptosis, we suppose that STS-induced cytosolic ATP increase is not related to PKC but associated with the induction of apoptotic process.
Under the experimental conditions used, cytosolic ATP was maintained at a level higher than the control value (of around 1.3 mM) for over 70 min. For HeLa cells incubated in culture medium, the STS-induced peak response was found to extend for up to 4 h after STS stimulation. Subsequently, the cytosolic ATP level gradually decreased, but was still higher than the control for 4–6 h in the time period during which activation of caspase-3 and internucleosomal DNA fragmentation were observed to be taking place. A similar long-lasting cytosolic ATP increase was observed also for cell death induced by TNFα/CHX. Although the time course of the decrease in cytosolic ATP was found to correlate with the time course of cell death, the percentage of reduction of the ATP signal was less prominent than that of cell death, suggesting the ATP level of living cells was still higher than the normal level. Therefore, it appears that cells undergoing apoptosis have an excess of cytosolic ATP throughout the deathbed phase where caspases are activated and DNA laddering is occurring.
Here, we detected a significant rise of the total ATP of lysates of nonluciferase-transfected HeLa cells after 30-min stimulation with STS. In contrast, a number of earlier studies showed that the level of cell lysate ATP decreases13, 15, 17, 18, 19 or remains unchanged5, 16, 20 after apoptotic stimulation for 1–16 h. It must be noted that the ATP level in the living cells, if measured in the very late phase of apoptotic process, may have been underestimated because of contamination by dead cells that had already lost ATP. In any case, changes in the cell lysate ATP do not always reflect changes in the cytosolic ATP level. Here, monitoring the bioluminescence reaction of luciferase expressed in the cytosol, for the first time, we could consistently observe a cytosolic ATP rise in intact HeLa, PC12 and U937 cells after apoptosis induction by STS, and in HeLa cells stimulated with etoposide or TNFα.
In a previous study, we have shown that AVD is a major hallmark of the apoptotic process.23 Such reduction in cell volume could by itself increase the cytosolic ATP concentration by condensation. However, the absolute degree of cell shrinkage was only about 10% during first hour of STS treatment,23 which cannot explain approx. 2.3-fold rise in the luminescence signal observed during this time in the present study. Moreover, STS-induced ATP rise was not inhibited by a K+ channel blocker (Ba2+), which also blocks AVD.23
The ATP response produced by an inducer of mitochondrion-mediated apoptosis (STS) was found to take place with a much earlier time course and a much greater amplitude than the response produced by an inducer of death receptor-mediated apoptosis (TNFα). An inducer of DNA damage-mediated apoptosis (etoposide) is known to stimulate mitochondria via caspase-2.29, 30 On the other hand, it is known that death receptor stimulation slowly induces secondary stimulation of mitochondria via Bid.31 These facts could suggest that mitochondrial stimulation is involved in the ATP response to apoptosis inducers. However, the STS-induced ATP rise itself was due mainly to stimulation of nonmitochondrial glycolytic reactions, and not of mitochondrial oxidative phosphorylation. It is therefore possible that some factor released from stimulated mitochondria is involved in enhancement of glycolysis. A possible candidate factor could be a proton, since it is known that STS-induced apoptosis is associated with an early cytosolic acidification, and oligomycin and protonophores suppress this effect as well as the following cell death.26 Further studies are required to test this possibility and to identify the factor. Mitochondrial ATP production in cells undergoing apoptosis might also play a supporting role in the maintenance of elevated cytosolic ATP levels, because STS-induced ATP responses became smaller and less prolonged in the presence of oligomycin. Such a role for mitochondrial ATP production is in line with recent reports that the mitochondrial membrane potential and ATP-generating function remain intact even after cytochrome c is released and caspase activation begins.16, 18, 32
In the present study, we used 2DG in glucose-free Ringer solution to inhibit glycolytic reactions and a mitochondrial Fo, F1-ATPase inhibitor, oligomycin, to suppress mitochondrial respiration. Since these maneuvers are strong assaults upon the cellular physiological functions and may have some nonspecific side effects, which may in turn modify cell sensitivity to the apoptosis-inducing agents, in the present study metabolic inhibitors were washed out after a short-time application. Thus, these maneuvers did not induce increases in the PI-positive cells under the present experimental conditions. Therefore, it is likely that execution of apoptosis took place in the conditions where glycolysis and mitochondrial respiration were fully recovered, and elimination of ATP rise only halted the initiation step of apoptosis without leading to necrosis.
The most notable findings in the present study are that inhibition of elevation of the cytosolic ATP level significantly suppressed Annexin V binding induced by STS or TNFα and prevented activation of caspase-3 and internucleosomal DNA fragmentation induced by STS. Even under conditions in which cytosolic ATP was maintained at a normal level, these execution events in apoptosis (caspase activation and DNA ladder) were abolished. Thus, maintaining cytosolic ATP at a normal level would be a useful method for protecting not only against necrotic cell death but also against apoptotic cell death associated with pathological insults such as ischemia/reperfusion, in which both forms of cell death are simultaneously induced.
Materials and Methods
Cell culture and establishment of luciferase-expressing cells
All cells were cultured on coverslips. HeLa cells were cultured in MEM culture medium, PC12 and HEK293T cells in DMEM medium, and U937 cells in RPMI 1640 medium. All culture media were supplemented with 10% FBS.
Since firefly luciferase introduced into cells by heterologous expression has been successfully used to continuously monitor the cytosolic ATP concentration,24, 28 we generated a HeLa cell line stably transfected with firefly luciferase.9 After selection, luciferase-expressing HeLa cells were maintained in DMEM containing 10% FCS, 500 μg/ml G418 and 100 μg/ml hygromycin. They were seeded onto 9 mm glass coverslips and grown to confluence (1–4 days) prior to luminescence measurements. Immunostaining showed that the whole cytosolic space was reactive to anti-luciferase antibody (Promega, Madison, WI, USA) (data not shown).
For transient expression of cytosolic luciferase in U937 and PC12 cells, the pGL3 control vector (Promega) was used. Cells were transiently transfected using Effectene transfection reagent (Qiagen, Valencia, CA, USA) according to the manufacturer's instructions.
Cytosolic ATP measurements
Measurements of cytosolic ATP were performed with a modification of a method used elsewhere,24 as described previously.9 Briefly, each coverslip, with around 1.6 × 105 HeLa or PC12 cells expressing luciferase, was washed with normal Ringer solution of the following composition (mM): 135 NaCl, 5 KCl, 2 CaCl2, 1 MgCl2, 5 Na-HEPES, 6 HEPES, 5 glucose (pH 7.4, 290 mosmol/kg H2O). The cells were then placed in a transparent 2 ml plastic cup (inner diameter 10 mm) containing 0.5 ml of normal Ringer solution. The cup was lowered directly into the thermostated recording chamber of the luminometer (Lumi-Counter 2500, Microtec, Chiba, Japan), which was coupled to a Windows-based PC. For luminescence measurements from U937 cells, a cell suspension containing 3 × 105 cells was centrifuged at 5000 r.p.m. for 1.5 min and resuspended into 0.5 ml of Ringer solution in a 2 ml plastic cup. After equilibration (5 min), the baseline luminescence (100–250 photons/10 s) was measured, and this was later subtracted for calculations of the percentage of luminescence changes and of the ATP concentrations. Luciferin (100 μM) was thereafter added from the 10 mM stock solution made up in 10 mM HEPES buffer. The luminescence was continuously monitored by integrating the photon counts over 10 s intervals at 37°C. The sample was rotated with a frequency of approx. 100 r.p.m. with alternation of the direction every 3–5 s to ensure proper mixing of the solutions. After the signal reached a steady-state level (10–15 min), apoptotic inducers (STS, TNFα+CHX, etoposide) or other reagents were added from 100 × −1000 × stock solutions. In all cases, at the concentrations used, these reagents did not have any direct effects on the luciferin–luciferase reaction. Continuous recordings were not possible in the culture medium because of interference with the luciferin–luciferase reaction by some components, such as dyes and serum. Thus, in some experiments (for Figure 2 data except for open circles and filled squares, and for Figure 5 data), cytosolic ATP measurements were carried out for HeLa cells, which were pretreated with staurosporine (STS) or TNFα (+CHX) for 25 or 60 min, respectively, in the absence or presence of metabolic inhibitors after incubating in culture medium for a specified length of time.
In order to relate the observed luminescence signals to cytosolic ATP concentrations, calibration procedures were performed according to Kennedy et al.24 In brief, HeLa cells were bathed in a cytosol-approximating electrolyte solution consisting of (mM): 10 NaCl, 20 KCl, 110 K aspartate, 0.5 MgCl2, 1.7 CaCl2, 5 EGTA, 10 HEPES (pH 7.2, pCa 7). The cells were then permeabilized by 2 μM digitonin, and varying concentrations of MgATP were added from a stock solution containing 100 mM Na2ATP and 100 mM MgSO4 in the above solution, and having a pH adjusted to 7.2 with KOH. The concentration–response curve thus obtained could be fitted to a Michaelis–Menten equation with a Km of 1.55±0.15 mM. The Km value is close to that observed for MIN6 cells transfected with luciferase.24
Total ATP measurements for cell extracts
Total ATP was quantified using a commercially available luciferin–luciferase assay kit (AF-2L1, DKK-TOA, Tokyo, Japan). Nontransfected HeLa cells on coverslips were washed with 0.5 ml Ringer solution. Then, the coverslips were placed in a 24-well plate with 300 μl of ATP-releasing reagent (AF-2K2, DKK-TOA). The plate was swirled gently, and lysis was allowed to proceed for 3.5 min at room temperature. The lysate was stirred with a micropipette, and a 50 μl aliquot of lysate was added to 400 μl Ringer solution. Then, luminescence was measured at 37°C by adding a 50 μl aliquot of luciferin–luciferase mixture from the ATP assay kit.
Annexin V-binding, caspase-3, DNA fragmentation and cell viability assays
Assays for Annexin V binding and PI staining were performed by using Annexin V–FITC Apoptosis Detection Kit (Sigma, St. Louis, MO, USA) according to the manufacturer's instructions. The cells were visualized under a fluorescence microscope, the digital images were transferred to the computer, and positive cells were counted manually inside a square area of 833 × 625 μm2, which contained about 1000 cells.
Caspase-3 activity was measured using a fluorometric assay,33 as described previously.23 In brief, the difference in fluorescence in the absence and in the presence of a specific inhibitor of caspase-3 was observed. The fluorogenic substrate for caspase-3 (Ac-DEVD-AMC), which was labeled with the fluorochrome 7-amino 4-methyl coumarin, and the tetrapeptide inhibitor of caspase-3 (Ac-DEVD-CHO) were provided in the CaspASE Assay System (Promega).
Internucleosomal DNA fragmentation was detected as DNA laddering, as reported previously.23, 34 Briefly, cells were digested in lysis buffer (10 mM EDTA/0.5% Na-N-lauroylsarcosinate/500 μg/ml RNase/50 mM Tris-HCl, pH 7.8) for 1 h at 37°C and treated with 500 μg/ml proteinase K for 1 h at 37°C. The chromosomal DNA was analyzed by agarose gel electrophoresis (2%) followed by staining with ethidium bromide.
Viability of cells was assessed using the Cell Counting Kit (Dojindo, Kumamoto, Japan) according to the manufacturer's instructions. This kit measures mitochondrial dehydrogenase activity using a colorimetric 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) assay, and is based on the principle that viable cells (but not dead cells) can reduce MTT.35
Chemicals
Luciferin was obtained from Promega, digitonin from Nacalai Tesque (Kyoto, Japan), CHX from Wako (Osaka, Japan), TNFα from Endogen (Woburn, MA, USA) and other chemicals from Sigma (St. Louis, MO, USA).
Statistics
Data are given as means±S.E.M. Comparisons between two experimental groups were made using the unpaired Student's t-test. Differences were considered to be statistically significance at P<0.05.
Abbreviations
- STS:
-
staurosporine
- TNFα:
-
tumor necrosis factor α
- CHX:
-
cycloheximide
- MTT:
-
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide
- DMSO:
-
dimethyl sulfoxide
- 2DG:
-
2-deoxy-D-glucose
References
Steller H (1995) Mechanisms and genes of cellular suicide. Science 267: 1445–1449
Thompson CB (1995) Apoptosis in the pathogenesis and treatment of disease. Science 267: 1456–1462
Kass GEN, Eriksson JE, Weis M, Orrenuis S and Chow SC (1996) Chromatin condensation during apoptosis requires ATP. Biochem. J. 318: 749–752
Richter C, Schweizer M, Cossarizza A and Franceschi C (1996) Control of apoptosis by the cellular ATP level. FEBS Lett. 378: 107–110
Leist M, Single B, Castoldi AF, Kuhnle S and Nicotera P (1997) Intracellular adenosine triphosphate (ATP) concentration: a switch in the decision between apoptosis and necrosis. J. Exp. Med. 185: 1481–1486
Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES and Wang X (1997) Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell 91: 479–489
Nicotera P, Leist M and Ferrando-May E (1998) Intracellular ATP, a switch in the decision between apoptosis and necrosis. Toxicol. Lett. 102-103: 139–142
Hu Y, Benedict MA, Ding L and Nunez G (1999) Role of cytochrome c and dATP/ATP hydrolysis in Apaf-1-mediated caspase-9 activation and apoptosis. EMBO J. 18: 3586–3595
Barros LF, Kanaseki T, Sabirov R, Morishima S, Castro J, Bittner CX, Maeno E, Ando-Akatsuka Y and Okada Y (2003) Apoptotic and necrotic blebs in epithelial cells display similar neck diameters but different kinase dependency. Cell Death Differ. 10: 687–697
Chou CC, Lam CY and Yung BYM (1995) Intracellular ATP is required for actinomycin D-induced apoptotic cell death in HeLa cells. Cancer Lett. 96: 181–187
Eguchi Y, Shimizu S and Tsujimoto Y (1997) Intracellular ATP levels determine cell death fate by apoptosis or necrosis. Cancer Res. 57: 1835–1840
Ferrari D, Stepczynska A, Los M, Wesselborg S and Schulze-Osthoff K (1998) Differential regulation and ATP requirement for caspase-8 and caspase-3 activation during CD95- and anticancer drug-induced apoptosis. J. Exp. Med. 188: 979–984
Lieberthal W, Menza SA and Levine JS (1998) Graded ATP depletion can cause necrosis or apoptosis of cultured mouse proximal tubular cells. Am. J. Physiol. 274: F315–F327
Leist M, Saingle B, Naumann H, Fava E, Simon B, Kuhnle S and Nicotera P (1999) Inhibition of mitochondrial ATP generation by nitric oxide switches apoptosis to necrosis. Exp. Cell Res. 249: 396–403
Nomura K, Imai H, Koumura T, Arai M and Nakagawa Y (1999) Mitochondrial phospholipid hydroperoxide glutathione peroxidase suppresses apoptosis mediated by a mitochondrial death pathway. J. Biol. Chem. 274: 29294–29302
Bossy-Wetzel E, Newmeyer DD and Green DR (1998) Mitochondrial cytochrome c release in apoptosis occurs upstream of DEVD-specific caspase activation and independently of mitochondrial transmembrane depolarization. EMBO J. 17: 37–49
Vander Heiden MG, Chandel NS, Schumacker PT and Thompson CB (1999) Bcl-xL prevents cell death following growth factor withdrawal by facilitating mitochondrial ATP/ADP exchange. Mol. Cell 3: 159–167
Waterhouse NJ, Goldstein JC, Von Ahsen O, Schuler M, Newmeyer DD and Green DR (2001) Cytochrome c maintains mitochondrial transmembrane potential and ATP generation after outer mitochondrial membrane permeabilization during the apoptotic process. J. Cell Biol. 153: 319–328
Gleiss B, Gogvadze V, Orrenius S and Fadeel B (2002) Fas-triggered phosphatidylserine exposure is modulated by intracellular ATP. FEBS Lett. 519: 153–158
Budd SL, Tenneti L, Lishnak T and Lipton SA (2000) Mitochondrial and extramitochondrial apoptotic signaling pathways in cerebrocortical neurons. Proc. Natl. Acad. Sci. USA 97: 6161–6166
Rizzuto R, Bernardi P and Pozzan T (2000) Mitochondria as all-round players of the calcium game. J. Physiol. (Lond.) 529: 37–47
Rutter GA and Rizzuto R (2000) Regulation of mitochondrial metabolism by ER Ca2+ release: an intimate connection. Trends Biochem. Sci. 25: 215–221
Maeno E, Ishizaki Y, Kanaseki T, Hazama A and Okada Y (2000) Normotonic cell shrinkage because of disordered volume regulation is an early prerequisite to apoptosis. Proc. Natl. Acad. Sci. USA 97: 9487–9492
Kennedy HJ, Pouli AE, Ainscow EK, Jouaville LS, Rizzuto R and Rutter GA (1999) Glucose generates sub-plasma membrane ATP microdomains in single islet beta-cells. Potential role for strategically located mitochondria. J. Biol. Chem. 274: 13281–13291
Gribble FM, Loussouarn G, Tucker SJ, Zhao C, Nichols CG and Ashcroft FM (2000) A novel method for measurement of submembrane ATP concentration. J. Biol. Chem. 275: 30046–30049
Matsuyama S, Llopis J, Deveraux QL, Tsien RY and Reed JC (2000) Changes in intramitochondrial and cytosolic pH: early events that modulate caspase activation during apoptosis. Nat. Cell Biol. 2: 318–325
DeLuca M (1976) Firefly luciferase. Adv. Enzymol. 44: 37–68
Maechler P, Wang H and Wollheim CB (1998) Continuous monitoring of ATP levels in living insulin secreting cells expressing cytosolic firefly luciferase. FEBS Lett. 422: 328–332
Robertson JD, Enoksson M, Suomela M, Zhivotovsky B and Orrenius S (2002) Caspase-2 acts upstream of mitochondria to promote cytochrome c release during etoposide-induced apoptosis. J. Biol. Chem. 277: 29803–29809
Lassus P, Opitz-Araya X and Lazebnik Y (2002) Requirement for caspase-2 in stress-induced apoptosis before mitochondrial permeabilization. Science 297: 1352–1354
Zhao Y, Li S, Childs EE, Kuharsky DK and Yin X-M (2001) Activation of pro-death Bcl-2 family proteins and mitochondria apoptosis pathway in tumor necrosis factor-alpha-induced liver injury. J. Biol. Chem. 276: 27432–27440
Bradbury DA, Simmons TD, Slater KJ and Crouch SPM (2000) Measurement of the ADP : ATP ratio in human leukaemic cell lines can be used as an indicator of cell viability, necrosis and apoptosis. J. Immunol. Methods 240: 79–92
Thornberry NA (1994) Interleukin-1 beta converting enzyme. Methods Enzymol. 244: 615–631
Shiokawa D, Ohyama H, Yamada T, Takahashi K and Tanuma S (1994) Identification of an endonuclease responsible for apoptosis in rat thymocytes. Eur. J. Biochem. 226: 23–30
Mosmann T (1983) Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J. Immunol. Methods 65: 55–63
Acknowledgements
This work was supported by the Grants-in-Aid for Scientific Research (A) and (C) from the Ministry of Education, Culture, Sports, Science and Technology of Japan (to YO and RZS). We acknowledge a visiting Professorship from the Japanese Government to MVZ We thank EL Lee for manuscript preparation, K Shigemoto and M Ohara for technical assistance, and T Okayasu for secretarial assistance.
Author information
Authors and Affiliations
Corresponding author
Additional information
Edited by DR Green
Rights and permissions
About this article
Cite this article
Zamaraeva, M., Sabirov, R., Maeno, E. et al. Cells die with increased cytosolic ATP during apoptosis: a bioluminescence study with intracellular luciferase. Cell Death Differ 12, 1390–1397 (2005). https://doi.org/10.1038/sj.cdd.4401661
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.cdd.4401661
Keywords
This article is cited by
-
Quantifying requirements for mitochondrial apoptosis in CAR T killing of cancer cells
Cell Death & Disease (2023)
-
Transcriptomic and biochemical analyses revealed antifungal mechanism of trans-anethole on Aspergillus flavus growth
Applied Microbiology and Biotechnology (2023)
-
Blocking GSDME-mediated pyroptosis in renal tubular epithelial cells alleviates disease activity in lupus mice
Cell Death Discovery (2022)
-
Arylamine N-acetyltransferase 1 deficiency inhibits drug-induced cell death in breast cancer cells: switch from cytochrome C-dependent apoptosis to necroptosis
Breast Cancer Research and Treatment (2022)
-
Inhibition of noncaspase proteases, calpain and proteasome, via ALLN and Bortezomib contributes to cell death through low degradation of pro-/anti-apoptotic proteins and apoptosis induction
Medical Oncology (2022)
