
Abstract
Bcl-xS, a proapoptotic member of the Bcl-2 protein family, is localized in the mitochondria and induces apoptosis in a caspase- and BH3-dependent manner by a mechanism involving cytochrome c release. The way in which Bcl-xS induces caspase activation and cytochrome c release, as well as the relationship between Bcl-xS and other proapoptotic members of the Bcl-2 family, is not known. Here we used embryonic fibroblasts derived from mice deficient in the multidomain proapoptotic members of the Bcl-2 family (Bax and Bak) and the apoptotic components of the apoptosome (Apaf-1 and caspase-9) to unravel the cascade of events by which Bcl-xS promotes apoptosis. Our results show that Bak but not Bax is essential for Bcl-xS-induced apoptosis. Bcl-xS induced activation of Bak, which in turn promoted apoptosis by apoptosome-dependent and -independent pathways. These findings provide the first evidence that a proapoptotic Bcl-2 family protein induces apoptosis exclusively via Bak.
Similar content being viewed by others

Introduction
Programmed cell death, or apoptosis, is a regulated mechanism by which unwanted cells are selectively eliminated. Key participants in apoptosis are intracellular organelles, the best characterized of which are the mitochondria. When, as a result of apoptotic stimulation, the outer mitochondrial membrane (OMM) becomes permeable, proteins (such as cytochrome c) that are normally stored in the intermembrane space are released to the cytosol. Once in the cytosol, cytochrome c induces generation of the oligomeric complex termed the ‘apoptosome’, which contains cytochrome c, Apaf-1, and caspase-9. Generation of the apoptosome leads to activation of caspase-9, which then cleaves and activates the effector caspases, caspase-3 and caspase-7 (for reviews, see Wang1 and Lim et al.2).
The apoptosome plays an important role in many apoptotic pathways, as shown for example by the resistance of cells deficient in Apaf-1, caspase-9, or caspase-3 to different apoptotic stimuli.3 This mitochondria-mediated apoptotic pathway is regulated by the critical regulators of apoptosis, the Bcl-2 family of proteins, which contains both anti- and proapoptotic members. The antiapoptotic members, such as Bcl-2 and Bcl-xL, protect cells from apoptosis and contain characteristic regions of Bcl-2 homology (BH) domains, designated BH1, BH2, BH3, and BH4. The proapoptotic members of the family can be divided into two subgroups: (1) proteins that contain two or three BH domains, such as Bax, Bak, and Bok, and (2) proteins such as Bad, Bid, Bik, Bim, Blk, Hrk, Noxa, and Puma, which contain only BH3, the domain essential for their binding to the antiapoptotic members of the family and for their killing effect (for reviews, see Gross et al.4, Borner5, Bouillet and Strasser,6 and Adams and Cory7). Experiments with cells from mice lacking both Bax and Bak [Bax−/− Bak−/− double knockout (DKO) mice] demonstrate that Bax and Bak have a role distinct from that of the BH3-only protein. Thus, for example, mouse embryonic fibroblasts (MEFs) lacking both Bax and Bak are resistant to apoptosis induced by overexpression of BH3-only proteins such as Bid, Bim, Bad, and Noxa, indicating that the BH3-only proteins require the presence of Bax/Bak proteins for their apoptotic effect.8, 9, 10 In addition, Bax and Bak can compensate for each other, as MEFs that express either Bax or Bak are sensitive to apoptosis induced by expression of BH3-only proteins.8, 9 In healthy cells, Bax usually exists as a monomer in the cytoplasm or loosely attached to the OMM, whereas Bak appears to be attached to the mitochondria. After apoptotic stimulation, the mitochondrial Bak undergoes conformational changes from an inactive to an active form, whereas Bax, in some cases, first translocates to the mitochondria and only then undergoes conformational changes to its active form. The Bak/Bax conformational changes include induction of an open conformation in which the N terminus (NT) of the proteins is exposed. This open conformation leads in turn to oligomerization of the nonactivated conformers, causing a cascade of Bax/Bak auto-oligomerization (for reviews, see Sharpe et al.11 and Degli Esposti and Dive12).
Bcl-xS, a proapoptotic protein of the Bcl-2 protein family, is an alternative splicing form of the Bcl-x gene. It contains the BH3 and BH4 domains and a transmembrane region.13 A number of studies have suggested that Bcl-xS might play an important role in some apoptotic systems. For example, expression of Bcl-xS is increased during apoptosis in transient forebrain global ischemia,14, 15 in involuting mammary epithelial cells,16 in hypertensive nephrosclerosis,17 and during apoptosis induced by balloon injury in carotid arteries18 or by interleukin-18 in endothelial cells.19 Bcl-xS was also suggested to participate in apoptosis of cancer cells, implying that targeting of Bcl-xS expression on cancer cells might be a useful therapeutic approach for the specific killing of these cells. These suggestions were supported by the findings that short duration of relapse-free survival and overall survival in acute myelogenous leukemia correlates with loss of Bcl-xS expression20 and that Bcl-xS plays a role in apoptosis induced by actinomycin D in human gastric cancer TMK-1 cells.21 Furthermore, infection with an adenoviral vector containing Bcl-xS cDNA specifically induces apoptosis in several types of cancer cells,22, 23, 24, 25 and transfer of Bcl-xS plasmid is effective in preventing and inhibiting rat hepatocellular carcinoma induced by N-nitrosomorpholine.26 Unraveling the mechanism of Bcl-xS-induced apoptosis could therefore have important clinical implications.
Studies by our group and others have shown that caspases are needed for the apoptotic effect of Bcl-xS and that ectopically expressed Bcl-xS is localized in the mitochondria, where it induces caspase-dependent and -independent cytochrome c release. Furthermore, Bcl-xS induces apoptosis in a BH3-dependent manner and the antiapoptotic proteins of the Bcl-2 family inhibit Bcl-xS-induced apoptosis.27, 28, 29, 30, 31, 32 Not yet known, however, are the initial events by which Bcl-xS induces mitochondrial dysfunction, the relationship between Bcl-xS and other proapoptotic members of the Bcl-2 family, or the role of the apoptosome in Bcl-xS-induced apoptosis. In an attempt to unravel these unknown aspects of Bcl-xS activity, we studied the mechanism of Bcl-xS-induced apoptosis in MEFs derived from mice deficient in distinct apoptotic components, including proteins of the apoptosome pathway and proapoptotic members of the Bcl-2 family. Our results showed that Bcl-xS induced apoptosis in these cells via Bak activation and, moreover, that the Bcl-xS-induced apoptosis was entirely dependent on Bak but not on Bax. In addition, we showed that Bcl-xS-induced apoptosis was mediated via apoptosome-dependent and -independent pathways. These findings provide the first evidence that apoptotic stimuli can promote apoptosis via a pathway in which Bak cannot be compensated for by Bax.
Results
The apoptosome is required for cell death induced by Bcl-xS
Transient cotransfection of wild-type (WT) MEFs with FLAG-Bcl-xS and secreted alkaline phosphatase (SEAP) expression vectors, and determination of cell viability by monitoring SEAP activity in the transfected cells 24 h later, revealed that Bcl-xS is capable of inducing cell death in MEFs in a dose-dependent manner (data not shown). This finding established that MEFs are suitable as a model for studying the molecular mechanism of Bcl-xS-induced apoptosis.
Previous studies from our laboratory showed that in many cell types, Bcl-xS induces apoptosis by a mechanism that involves cytochrome c release30 and caspase activation.28, 30 Those results thus suggested that the Bcl-xS-induced apoptosis is mediated via the apoptosome, which in turn might lead to caspase activation. To substantiate this notion, we examined the ability of Bcl-xS to induce apoptosis in MEFs derived from mice deficient in the apoptosome components Apaf-1 or caspase-9.
FLAG-Bcl-xS and SEAP expression vectors were cotransfected into MEFs deficient in Apaf-1 or caspase-9 or into their corresponding WT MEFs (WT1 for Apaf-1-deficient and WT2 for caspase-9-deficient MEFs). We assessed cell viability by monitoring SEAP activity in the cells 24, 48, 72, and 96 h after transfection. Figure 1a shows that, at all time points examined, both Apaf-1-deficient and caspase-9-deficient MEFs were much more resistant to Bcl-xS-induced cell death than their corresponding WT MEFs. The resistance of Apaf-1−/− and caspase-9−/− MEFs to Bcl-xS-induced apoptosis could not be attributed to the absence or decline of Bcl-xS expression in these cells, as Bcl-xS protein was expressed in comparable amounts in the different Bcl-xS-transfected MEFs (Figure 1b). Because the SEAP activity assay reflects the effect of Bcl-xS in a pool of cells rather than on a single-cell basis, and to exclude the possibility that deficiency of Apaf-1 or caspase-9 does not prevent Bcl-xS-induced cell death but only delays it, we also assessed cell viability by the colony-formation assay, which assesses the effect of the transfected gene both over long periods and on a single-cell basis. The puromycin-resistant plasmid, pGKpuro, was cotransfected with either FLAG-Bcl-xS or empty vector into MEFs deficient in Apaf-1 or caspase-9 or into their corresponding WT MEFs, and the numbers of puromycin-resistant colonies were counted 2 weeks later. As shown in Figure 1c, the percentage of puromycin-resistant colonies obtained in FLAG-Bcl-xS-transfected cultures from cultures transfected with the empty vector was much higher in MEFs deficient in Apaf-1 or caspase-9 than in their corresponding WT MEFs. Taken together, these results suggest that Apaf-1 and caspase-9 are required for Bcl-xS-induced cell death, and therefore that the apoptosome plays an important role in execution of the apoptotic effect of Bcl-xS. Because the apoptosome induces caspase activation, these results further suggest that caspases might play an important role in Bcl-xS-induced apoptosis in MEFs. To substantiate this notion, we examined the ability of Z-VAD-FMK, a broad-spectrum inhibitor of caspases, to inhibit Bcl-xS-induced apoptosis in WT MEFs. As shown in Figure 2a, Z-VAD-FMK substantially inhibited Bcl-xS-induced cell death.

Apaf-1 and caspase-9 are required for Bcl-xS-induced cell death. Apaf-1−/− and caspase-9−/−, and their corresponding WT MEFs (WT1 and WT2, respectively) were transiently cotransfected for 24 h with the reporter SEAP and FLAG-Bcl-xS expression vectors. (a) SEAP activity in each transfection was determined after 24, 48, 72, and 96 h, as described in Materials and Methods. Cell survival is defined as SEAP activity in cultures transfected with FLAG-Bcl-xS and is expressed as a percentage of SEAP activity in cultures transfected with the control vector pcDNA3. Data are expressed as mean values±S.D. (n=3). (b) Expression of FLAG-Bcl-xS in the different MEF cultures transfected with FLAG-Bcl-xS plasmid was assayed by immunoblot analysis, as described in Materials and Methods. The data shown are from a representative experiment that was also analyzed for cell viability, and its SEAP values are included in the mean values of the data presented for the 24 h time point in (a). (c) Apaf-1−/− and caspase-9−/−, and their corresponding WT MEFs were cotransfected with pGKpuro and either FLAG-Bcl-xS or pcDAN3 as described in Materials and Methods. Puromycin-resistant clones were counted 2 weeks after selection. Colony formation is defined as the number of puromycin-resistant clones obtained from cultures transfected with FLAG-Bcl-xS and is expressed as a percentage of puromycin-resistant clones in cultures transfected with the control vector pcDNA3. Data are expressed as mean values±S.D. (n=3). An asterisk denotes a significant difference in the percentage of cell survival between Apaf-1−/− or caspase-9−/− MEFs and their corresponding WT MEFs (P<0.02, two-tailed Student's t-test)

Bak is required for Bcl-xS-induced cell death. Bax−/−, Bak−/−, and DKO, and their corresponding WT MEFs (WT) were transiently cotransfected with the reporter SEAP and FLAG-Bcl-xS expression vectors. Z-VAD-FMK (100 μM) (Z-VAD) was added to some of the cultures as described in Materials and Methods. (a) SEAP activity in each transfection was determined after 24, 48, 72, and 96 h, as described in Materials and Methods. Cell survival is defined as SEAP activity in cultures transfected with FLAG-Bcl-xS and is expressed as the percentage of SEAP activity in cultures transfected with the control vector pcDNA3. Data are expressed as mean values±S.D. (n=3). (b) Expression of FLAG-Bcl-xS was assayed in the different FLAG-Bcl-xS MEFs transfected cultures, as described in Figure 1. The data shown are from a representative experiment that was also analyzed for cell viability, and its SEAP values are included in the mean values of the data presented for the 24 h time point in (a). (c) Bax−/−, Bak−/−, and DKO, and their corresponding WT MEFs (WT) were cotransfected with pGKpuro and either FLAG-Bcl-xS or pcDAN3 as described in Materials and Methods, and the colony-formation data were expressed as described in Figure 1. An asterisk denotes a significant difference in the percentage of cell survival between Bak−/− or DKO MEFs and WT MEFs (P<0.002, two-tailed Student's t-test)
Bcl-xS-induced apoptosis is mediated by a Bak-dependent pathway that cannot be compensated for by Bax
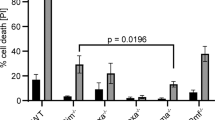
Studies have shown that BH3-only proteins, as well as various other apoptotic stimuli, require the presence of Bax and Bak proteins for their apoptotic effect, and that each of these proteins can compensate for the other.8, 9 To determine whether Bax and/or Bak are needed for cell death induced by Bcl-xS, we cotransfected FLAG-Bcl-xS and SEAP expression vectors into Bax−/−, Bak−/−, DKO, or WT MEFs, and assessed cell viability by monitoring SEAP activity in the transfected cells 24, 48, 72, and 96 h later. Figure 2a shows that WT MEFs and Bax-deficient MEFs were highly susceptible to Bcl-xS-induced cell death, whereas both Bak-deficient and DKO MEFs were much more resistant at all time points tested. The resistance of Bak−/− and DKO MEFs to the Bcl-xS-induced cell death could not be attributed to the lack of or decline in Bcl-xS expression, as Bcl-xS protein was expressed in comparable amounts in the different Bcl-xS-transfected MEFs (Figure 2b). Furthermore, analysis of colony formation (Figure 2c) confirmed the results obtained by the SEAP assay and showed that both Bak-deficient and DKO MEFs were completely resistant to Bcl-xS-induced cell death, whereas WT MEFs and Bax-deficient MEFs were highly susceptible to it. These results thus suggest that Bak is required for Bcl-xS-induced apoptosis, but Bax is not.
Bcl-xS induces Bak activation in an Apaf-1-, caspase-9-, and Bax-independent manner and Bax activation in a Bak-dependent manner
The above results showed that Bcl-xS requires Bak but not Bax for its apoptotic effect. To further analyze the roles of Bak and Bax in Bcl-xS-induced apoptosis, we examined whether Bcl-xS induces Bak or Bax activation and the extent to which such activation, if it exists, depends on the presence of Bax, Bak, Apaf-1, or caspase-9. Previous studies have shown that after apoptotic stimulation, Bak and Bax undergo conformational changes from an inactive to an active form, a process that includes exposure of their NTs.33, 34 The exposed NT conformation of Bax or Bak can be recognized by specific antibodies directed against the NT region of these proteins.35, 36 Detection of Bax or Bak proteins by these specific antibodies shows that these proteins have an NT-exposed conformation, suggesting that they are activated. Therefore, in an attempt to examine the effect of Bcl-xS on Bak and Bax activation, the different MEFs were transfected with the green fluorescent protein (GFP) GFP-Bcl-xS expression vector, and exposure of the NTs of endogenous Bak or Bax proteins in the GFP-Bcl-xS-expressing cells was detected by immunofluorescence staining with anti-Bak-NT or anti-Bax-NT antibodies.
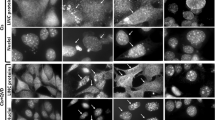
First we examined Bak activation. As exemplified in Figure 3a, expression of GFP-Bcl-xS in the WT MEFs induced apoptosis in the transfected cells, indicated by the fragmented morphology of nuclei of the GFP-Bcl-xS-expressing cells compared to the healthy nuclear morphology of neighboring cells not expressing GFP-Bcl-xS. Importantly, moreover, the GFP-Bcl-xS-expressing cells were stained with anti-Bak-NT antibodies, whereas the nontransfected cells were not. These results suggested that the expression of Bcl-xS in MEFs induced Bak activation. Examination of anti-Bak-NT staining in GFP-Bcl-xS-transfected Apaf-1−/−, caspase-9−/−, and Bax−/− MEFs revealed that, as in WT MEFs, the GFP-Bcl-xS-expressing cells were also stained with the anti-Bak-NT antibodies. This suggested that Bcl-xS had induced Bak activation in these cells as well, and that the activation was independent of Apaf-1, caspase-9, and Bax. The staining pattern of activated Bak in all of the MEFs was similar if not identical to the cellular distribution pattern of GFP-Bcl-xS, suggesting that these two proteins were colocalized. Since Bcl-xS is strongly associated with the mitochondria28, 37 these results further suggested that this is the colocalization site of the two proteins. Quantitative analysis of the Bak-NT-stained GFP-Bcl-xS-positive cells relative to the total number of GFP-Bcl-xS-expressing cells in WT, Apaf-1−/−, caspase-9−/−, or Bax−/− MEFs revealed that almost all of the GFP-Bcl-xS-expressing cells in these different MEFs (95±3, 88±5, 85±6, or 88±5%, respectively) contained activated Bak (Figure 3b). To exclude the possibility that the transfection procedure rather than Bcl-xS expression was responsible for Bak activation, we examined Bak-NT exposure in WT, Apaf-1−/−, caspase-9−/−, and Bax−/− MEFs transfected with GFP expression vector. Quantitative analysis (Figure 3b) of the Bak-NT-stained GFP-positive cells relative to the total number of GFP-expressing cells in these MEFs revealed that only a small proportion of the GFP-expressing cells (about 2–15%) contained activated Bak. These results thus demonstrate that Bak-NT exposure is a result of Bcl-xS expression and not of the transfection procedure.

Bcl-xS induces exposure of Bak-NT in an Apaf-1-, caspase-9-, and Bax-independent manner. WT, Apaf-1−/−, caspase-9−/−, or Bax−/− MEFs were transiently transfected with GFP-Bcl-xS or GFP expression vectors, as described in Materials and Methods. After 24 h, the cells [GFP-Bcl-xS transfected (a) and GFP transfected (not shown)] were stained with anti-Bak-NT antibody and with Hoechst 33258 (for staining of nuclei) and visualized by fluorescence microscopy using the appropriate filter sets, as described in Materials and Methods. The images of each MEF cell line represent the same field visualized separately for the detection of GFP-Bcl-xS expression, Bak-NT staining, and Hoechst-stained nuclei (Nuclei). The results presented are from a representative experiment (one of at least three experiments). Arrows indicate nuclei of cells that do not express Bcl-xS. (b) Quantitative analysis of GFP-Bcl-xS- or GFP-positive cells exhibiting Bak-NT immunoreactivity, expressed as a percentage of the total number of GFP-Bcl-xS- or GFP expressing cells in the different MEFs. Each histogram indicates mean values±S.D. of a total of at least 200 cells from at least three independent experiments
Next we examined the effect of Bcl-xS on Bax activation. GFP-Bcl-xS-expressing WT, Apaf-1−/−, and caspase-9−/− MEFs all showed positive staining with anti-Bax NT antibodies (Figure 4a), indicating that although Bax was not required for Bcl-xS-induced apoptosis in MEFs, it was nevertheless activated. This finding also showed that the Bcl-xS-induced activation of Bax was independent of Apaf-1 and caspase-9. In contrast to the effect of GFP-Bcl-xS on Bax activation observed in WT, Apaf-1−/−, and caspase-9−/− MEFs, it did not induce Bax activation in Bak−/− MEFs, as shown by the failure of these cells to exhibit immunoreactivity of Bax NT. These results thus suggest that the Bcl-xS-induced activation of Bax is Bak-dependent. In addition, as shown above for Bak, in all GFP-Bcl-xS-expressing MEFs, the activated Bax was colocalized with Bcl-xS, suggesting that the Bcl-xS-induced activated Bax is present in the mitochondria. Quantitative analysis (Figure 4b) showed that in WT and Apaf-1−/− MEFs the percentage of GFP-Bcl-xS-expressing cells exhibiting Bax activation was 91±6 and 89±9% respectively, whereas in Bak−/− MEFs it was only 3±5%. Interestingly, in caspase-9−/− MEFs, only 51±2% of the GFP-Bcl-xS-expressing cells exhibited Bax NT staining, indicating that Bax activation could be both dependent on caspase-9 and independent of it. Quantitative analysis (Figure 4b) of the Bax-NT-stained GFP-positive cells relative to the total number of GFP-expressing cells in WT, Apaf-1−/−, caspase-9−/−, or Bak−/− MEFs revealed that only a small proportion of the GFP-expressing cells (about 2–13%) contained activated Bax. These results thus demonstrate that Bax-NT exposure results from Bcl-xS expression and not from the transfection procedure.

Bcl-xS induces Bax activation in a Bak-dependent manner. WT, Apaf-1−/−, caspase-9−/−, or Bak−/− MEFs were transiently transfected with GFP-Bcl-xS or GFP expression vectors, as described in Materials and Methods. After 24 h, the cells [GFP-Bcl-xS transfected (a) and GFP transfected (not shown)] were stained with anti-Bax-NT antibody and with Hoechst 33258 (for staining of nuclei) and visualized by fluorescence microscopy using the appropriate filter sets, as described in Materials and Methods. The images of each MEF cell line represent the same field visualized separately for the detection of GFP-Bcl-xS expression, Bak-NT staining, and Hoechst-stained nuclei (Nuclei). The results presented are from a representative experiment (one of at least three experiments). (b) Quantitative analysis of GFP-Bcl-xS- or GFP-positive cells exhibiting Bax-NT immunoreactivity, expressed as a percentage of the total number of GFP-Bcl-xS- or GFP expressing cells in the different MEFs. Each histogram indicates mean values±S.D. of a total of at least 200 cells from at least three independent experiments. An asterisk denotes a significant difference in the percentage of cell survival between caspase-9−/− or Bak−/− MEFs and WT MEFs (P<0.0003, two-tailed Student's t-test)
Bcl-xS-induced cytochrome c release is dependent on Bak but independent of Apaf-1, caspase-9, and Bax
The experiments depicted in Figure 1a showed that Apaf-1 and caspase-9 are needed for Bcl-xS-induced apoptosis, suggesting a role for the apoptosome in Bcl-xS-induced cell death. Since generation of the apoptosome is induced by cytochrome c released from the mitochondria,38 we wanted to confirm that cytochrome c is indeed released from the mitochondria in Bcl-xS-induced apoptosis in MEFs and to examine the dependence of such release on expression of the apoptotic proteins Bax, Bak, caspase-9, and Apaf-1. The different MEFs were transfected with GFP-Bcl-xS and the cellular distribution of cytochrome c in the transfected cells was examined by immunofluorescence staining with anti-cytochrome c antibodies. As shown in Figure 5a, healthy WT MEFs that did not express GFP-Bcl-xS exhibited a scattered, punctuated cytochrome c staining pattern typical of the subcellular distribution of mitochondria, whereas the GFP-Bcl-xS-expressing MEFs exhibited a diffuse cytochrome c staining pattern suggestive of release of cytochrome c from the mitochondria. A similar diffuse staining pattern of cytochrome c was obtained in GFP-Bcl-xS-expressing Apaf-1−/−, caspase-9−/−, and Bax−/− MEFs. In contrast, GFP-Bcl-xS-expressing Bak−/− and DKO MEFs did not exhibit this diffuse staining pattern but rather the typical mitochondrial punctuated pattern, which was indistinguishable from that of the neighboring MEFs not expressing GFP-Bcl-xS. These results thus suggested that in Bak−/− or DKO MEFs, Bcl-xS expression could not induce the release of cytochrome c. Quantitative analysis showed that relative to the total number of GFP-Bcl-xS-expressing cells in each of the different MEFs, the percentage of GFP-Bcl-xS-expressing cells exhibiting cytochrome c release in WT, Apaf-1−/−, caspase-9−/−, Bax−/−, Bak−/−, and DKO MEFs was 77±7, 94±4, 92±4, 50±10, 5±3, and 15±0.7%, respectively (Figure 5b), and further showed that the Bcl-xS-induced cytochrome c release is mediated by a pathway that is dependent on Bak but independent of Apaf-1, caspase-9, and Bax. Quantitative analysis of GFP-expressing cells exhibiting cytochrome c release relative to the total number of GFP-expressing cells in WT, Apaf-1−/−, caspase-9−/−, Bax−/−, Bak−/−, and DKO MEFs revealed that only a small proportion of the GFP-expressing cells (about 2–14%) exhibited cytochrome c release. These results thus demonstrate that the cytochrome c release is a result of Bcl-xS expression and not of the transfection procedure.

Bcl-xS induces release of cytochrome c in a Bak-dependent manner. WT, Apaf-1−/−, caspase-9−/−, Bak−/−, Bax−/−, or DKO MEFs were transiently transfected with GFP-Bcl-xS or GFP expression vectors, as described in Materials and Methods. After 24 h, the cells [GFP-Bcl-xS transfected (a) and GFP transfected (not shown)] were stained with anti-cytochrome c (Cyt c) antibody and with Hoechst 33258 (for staining of nuclei) and visualized by fluorescence microscopy using the appropriate filter sets, as described in Materials and Methods. The images of each MEF cell line represent the same field visualized separately for the detection of GFP-Bcl-xS expression, Bak-NT staining, and Hoechst-stained nuclei (Nuclei). The results presented are from a representative experiment (one of at least three experiments). (b) Quantitative analysis of GFP-Bcl-xS- or GFP positive cells exhibiting cytochrome c release, expressed as a percentage of total GFP-Bcl-xS- or GFP-positive cells in the different MEFs. Each histogram indicates mean values±S.D. of a total of at least 200 cells from at least three independent experiments. An asterisk denotes a significant difference in the percentage of cells exhibiting cytochrome c release compared to WT MEFs (P<0.001, two-tailed Student's t-test). Note that that the difference between Bax−/− and WT was not significant (P>0.1; n=3, two-tailed Student's t-test)
Discussion
Four important conclusions can be drawn from the results of this study. First, apoptosis can be mediated entirely by Bak. Second, Bak can induce Bax activation (or at least exposure of its NT). Third, the mechanism of Bcl-xS-induced apoptosis depends on Bak activation, which is followed by multiple downstream pathways that culminate in apoptosome-dependent and -independent pathways. Fourth, compared to other BH3-only proteins, the mechanism of action of Bcl-xS is unique. These conclusions are based on the following findings: (1) Bak−/− and DKO (but not Bax−/−) SV40 transformed MEFs were resistant to both the short-term and the long-term apoptotic effects of Bcl-xS. (2) Bcl-xS induced Bak-NT exposure in WT MEFs as well as in MEFs deficient in Bax, caspase-9, or Apaf-1, suggesting that Bcl-xS induced Bak activation and that this activation was upstream of Bax, caspase-9, and Apaf-1. It should be noted that NT exposure in Bak does not demonstrate activation of this protein, but only suggests that it might occur. However, since we showed here that Bak was needed for Bcl-xS-induced apoptosis, and since others have shown that the apoptotic effects of Bak are mediated by activated Bak,34, 39 it seems reasonable to suggest that the Bcl-xS-induced apoptosis was mediated by activated Bak, and thus that the observed exposure of NT indeed represented Bak activation. (3) Bcl-xS expression induced exposure of Bax NT in WT MEFs, suggesting that Bcl-xS induced Bax activation as well. This exposure/activation, however, was completely blocked in Bak−/− MEFs but not in Apaf-1−/− or caspase-9−/− MEFs, suggesting that Bak was required for the Bcl-xS-induced activation of Bax. (4) The Bcl-xS-induced release of cytochrome c was dependent on Bak but not on Bax, Apaf-1, or caspase-9. (5) Apaf-1−/− and caspase-9−/− MEFs were partially resistant to Bcl-xS-induced cell death, suggesting that Bcl-xS induced cell death via both apoptosome-dependent and -independent pathways.
Role of Bax in Bcl-xS-induced apoptosis
Although our findings showed that Bax was not needed for Bcl-xS-induced apoptosis, they nevertheless also showed that Bcl-xS induced exposure of Bax-NT in a Bak-dependent manner. One interpretation of these results is that Bax is activated by a Bak-dependent mechanism and subsequently participates in the death process, albeit in a nonessential role. Alternatively, it is possible that the exposure of Bax-NT does not lead to full activation of Bax, and thus that Bax does not contribute to the Bcl-xS-induced death process. At present, we cannot distinguish between these possibilities. Interestingly, the Bcl-xS-induced exposure/activation of Bax-NT was observed in only 50% of the Bcl-xS-expressing caspase-9−/− MEFs, whereas in the WT and Apaf-1−/− MEFs almost all of the Bcl-xS-expressing cells exhibited Bax-NT exposure/activation. These results suggested that the Bak-dependent, Bcl-xS-induced exposure/activation of Bax-NT was mediated by both caspase-9 -dependent and -independent pathways. The finding of a Bax activation pathway that is dependent on caspase-9 but not on Apaf-1 provides the first reported evidence that caspase-9 can lead to Bax activation, and is in line with proposed models suggesting that BH3 proteins might, inter alia, activate initiator caspases upstream of the mitochondria.11 Previous studies by our groups showed that the mitochondria are the main target of Bcl-xS.27, 37 This finding together with the present findings that Bcl-xS induces cytochrome c release and is colocalized with activated Bak or Bax suggests that the main target of Bcl-xS-induced activated Bak and Bax is the mitochondria. However, Bax and Bak were also shown to be present and to play an apoptotic role in the endoplasmic reticulum (ER).40, 41 Furthermore, apoptotic crosstalk between the ER and mitochondria has been reported.42 Therefore, we cannot exclude the possibility that some of the Bcl-xS-induced Bak and Bax activation occurs in the ER. Activation of ER Bak and Bax might lead to release from the ER of apoptogenic agents, for example calcium, which in turn converge to the mitochondria and promote perforation of the OMM.
Role of the apoptosome in Bcl-xS-induced apoptosis
In a previous study, our group showed that Bcl-xS induces caspase-dependent apoptosis via a mechanism that involves cytochrome c release.30 Those findings thus suggested that the Bcl-xS-induced apoptosis is mediated via the apoptosome. The results presented here proved that the apoptosome plays an important role in the Bcl-xS-induced cell death. The death-inducing effect of the apoptosome is likely to be propagated by the apoptosome's target caspases, such as caspase-3 and -7. Notably, assay of SEAP activity revealed that both Apaf-1- and caspase-9-deficient MEFs were only partially resistant to Bcl-xS-induced cell death, whereas the colony assay revealed complete resistance to Bcl-xS-induced cell death by the Apaf-1-deficient MEFs. These variations might be attributable to the differences between these two assays, namely transient transfection (for 1–4 days) without selection compared to stable (14-day) transfection and selection for puromycin-resistant clones. In view of the fact that the partial resistance of Apaf-1-deficient MEFs to Bcl-xS-induced cell death was evidenced both by the SEAP activity assay (Figure 1a) and by estimation of the number of GFP-positive cells (data not shown), and because caspase-9-deficient MEFs were partially resistant to Bcl-xS-induced cell death (as shown by both the SEAP activity and the colony-formation assays), we cannot exclude the possibility that this cell death was also mediated by an apoptosome-independent pathway. The nature of the apoptosome-independent pathway is not known, but we suggest that it might be mediated via mitochondrial membrane perforation, which in addition to releasing cytochrome c can lead to release of the high-temperature requirement protein A2/Omi and of endonuclease G (for review, see Donovan and Cotter43), known to contribute to apoptosome-independent apoptosis. In addition, by demonstrating that the Bcl-xS-induced release of cytochrome c was dependent on Bak but independent of Apaf-1, caspase-9, and Bax, the present results also show that cytochrome c release occurs downstream of Bak activation and upstream of apoptosome activation. Figure 6 presents a model summarizing our results and conclusions.

A model depicting the apoptotic effects of Bcl-xS in MEFs. Expression of Bcl-xS in MEFs induces exposure of NT of Bak, leading to its activation. The activated Bak can induce three pathways: a major pathway leading to cytochrome c release, which in turn causes apoptosome activation and cell death in a caspase-dependent manner; a second pathway, which leads to Apaf-1- and caspase-9-independent cell death; and a third pathway, which induces the exposure of Bax NT, both in a caspase-9-dependent and -independent manner, causing its activation. The first and the second pathways contribute to the death process. The role of the third pathway is not yet known
Specificity of Bak activation
Studies have shown that DKO MEFs are resistant to diverse apoptotic stimuli, including overexpression of BH3-only proteins such as Bid, Bim, Bad, and Noxa. However, MEFs deficient only in Bax or Bak proteins (but not both) are susceptible to all of these apoptotic stimuli.8, 9 Those findings led to the hypothesis that apoptosis induced by multiple apoptotic stimuli, probably via the BH3-only proteins, requires the action of the multidomain proapoptotic proteins Bax and Bak. In this mechanism, each of the proapoptotic proteins can compensate for the absence of the other. Other studies have shown, however, that in some cases, apoptosis is promoted only by Bax. For example, neuronal primary cultures derived from Bax-deficient mice are resistant to apoptosis induced by overexpression of Bim, Hrk, or Bid,44 and Bax-deficient carcinoma cells are resistant to Bik-induced apoptosis.45
Our present results, which showed that the Bcl-xS-induced cell death depends only on Bak, provide the first indication that an apoptotic stimulus can be mediated by Bak, independently of Bax. Moreover, these results, together with the above-mentioned findings in connection with Bax, suggest that apoptotic stimuli such as those mediated by the BH3-only proteins might induce apoptosis via different mechanisms, some dependent entirely on a particular multidomain proapoptotic protein (e.g. Bax or Bak), and others that either require the combined action of several multidomain proapoptotic proteins or are mediated via one proapoptotic protein whose action can however be compensated by others in its absence. The finding that Bcl-xS requires Bak but not Bax to mediate its apoptotic effect also suggests that, compared to other BH3-only proteins such as Bid, Bim, and Bik, the mechanism of action of Bcl-xS is unique, and further suggests that it might participate in distinct apoptotic systems.
Mechanism of Bcl-xS-induced Bak activation
How does Bcl-xS induce Bak activation?
Bak is constitutively localized in the mitochondria. During apoptosis, Bak homo-oligomerizes, forms focal complexes on mitochondria, and exposes its NT epitope.8, 34 It was suggested that Bak utilizes mitochondrial-resident molecules to regulate its activation.12, 46 Thus, a mechanism whereby Bcl-xS induces Bak activation might require direct binding of Bcl-xS to Bak. This activation might be mediated by a direct effect of Bcl-xS on Bak or indirectly by displacement from Bak of inhibitors such as the antiapoptotic members of the Bcl-2 family, or proteins (such as VDAC2) that do not belong to the Bcl-2 family.47 With regard to the possibility of direct binding of Bcl-xS to Bak, it should be noted that such interaction could not be detected in yeast by the two-hybrid system.48 Alternatively, Bcl-xS might activate Bak indirectly by binding to inhibitors of Bak, thus preventing them from acting on Bak. It was shown, for example, that the antiapoptotic protein Bcl-xL is bound predominantly to Bak in most cultured cells, and that this binding inhibits Bak activation. Apoptotic stimuli weaken this interaction, leading to the release of Bak from Bcl-xL.34, 49 Since studies by our group and others have shown that Bcl-xS can bind to Bcl-xL,28, 32 it is possible that Bcl-xS acts by binding to Bcl-xL. Further studies are needed to determine which, if any, of these possible mechanisms promote Bcl-xS-dependent Bak activation.
Materials and Methods
Cell culture
WT and knockout transformed MEFs were grown in high-glucose Dulbecco's modified Eagle's medium (DMEM; Sigma, St. Louis, MO, USA) supplemented with 10% heat-inactivated fetal calf serum (Sigma). SV-40-transformed Bax−/−, Bak−/−, and DKO, and their corresponding WT MEFs were generously provided by Stanley J Korsmeyer (Harvard Medical School, Boston, MA, USA). Apaf-1−/−-transformed MEFs were generously provided by Tak W Mak (Advanced Medical Discovery Institute, University of Toronto, Toronto, Ontario, Canada). WT 129J/B6 primary MEFs corresponding to the Apaf-1−/− MEF genotype50 were obtained from Adi Kimchi (The Weizmann Institute of Science, Rehovot, Israel). These primary MEFs were immortalized by SV-40 transformation and designated WT1. Caspase-9−/− and the corresponding WT immortalized MEFs (designated WT2) were generously provided by Andreas Strasser (The Walter and Eliza Hall Institute of Medical Research, Parkville, Victoria, Australia). Caspase-9−/− and WT2 MEFs were immortalized by the 3T9 protocol.51
Generation of WT2 MEFs
WT 129J/B6 primary MEFs were transformed by transfection with SV-40 whole genome DNA (a gift from Atan Gross, The Weizmann Institute of Science) using the FuGENE 6 transfection reagent (Roche Diagnostics, Mannheim, Germany). At 1 day before transfection, the cells were seeded at a density of 2 × 105 cells per well in six-well plates. To each dish, we added 200 μl of DNA–FuGENE 6 mixture (1 μg DNA and 3 μl of FuGENE 6 reagent in 200 μl of OptiMEM (Invitrogen Life Technologies, Paisley, UK)) according to the manufacturer's instructions. The cells were split 24 h after transfection and seeded at 2 × 104 cells per 10 cm2 dish. A pool of transformed clones was collected 14 days after transfection for further experiments.
Plasmids
The FLAG-Bcl-xS, GFP-Bcl-xS, and pCMV-SEAP expression vectors used in this study were generated as previously described.28
Transfection
At 1 day before transfection, the different MEF cells were seeded at a density of 2 × 105 cells per well in six-well plates or in 35 mm dishes. To each well or dish, we added 1 ml of DNA–lipofectamine mixture (Invitrogen) (2 μg DNA and 10 μg lipofectamine in 1 ml of OptiMEM), according to the manufacturer's instructions. After incubation of the cells with the DNA–lipofectamine mixture for 5 h, DMEM supplemented with 10% serum without or with Z-VAD-FMK (ICN Biomedicals, Irvine, CA, USA) was added and incubation was continued. The transfection efficiencies of the different MEFs were estimated by the numbers of GFP-positive cells obtained after cotransfection of each of the MEFs with the empty vector pcDNA3 and GFP expression vector and the SEAP activity ratios between the different MEFs cotransfected with pcDNA3 and SEAP expression vectors. The transfection efficiencies (%) were found to be about 10, 8, 20, 8, 26, 26, 18, and 8 for WT, WT1, WT2, Bax−/−, Bak−/−, DKO, Apaf-1−/−, and caspase-9−/−, respectively. Ratios of the different DNA vectors were 3 : 1 : 2 for FLAG-Bcl-xS/pEGFP/SEAP plasmid or pcDNA3/pEGFP/SEAP plasmid in the transfection for SEAP cell survival assays; 1 : 3 for GFP-Bcl-xS/pcDNA3 or GFP/pcDNA3 in the transfection for immunofluorescence staining; and 1 : 5 : 10 : 4 for pGKpuro/pEGFP/FLAG-Bcl-xS/pcDNA3 or 1 : 5 : 14 for pGKpuro/pEGFP/pcDNA3 for the colony-formation assay.
Immunofluorescence staining
The different MEFs were grown in 35 mm dishes, 2 × 105 cells per dish, on 18 mm coverslips coated with collagen, and transfected as described above. After 24 h, the transfected cells were washed twice with buffer B [2 mM CaCl2 in Tris-buffered saline (TBS)], fixed with 4% paraformaldehyde for 30 min, washed twice with buffer B, and permeabilized with 0.1% Triton for 10 min. After two more washes with buffer B, the cells were incubated with buffer A (2 mM CaCl2, 2% bovine serum albumin in TBS), with the addition of normal goat IgG (200 μg/ml; Jackson ImmunoResearch Laboratories, West Grove, PA, USA) to block nonspecific binding. After being washed with buffer A, the cells were incubated for 1 h with buffer A containing 10 μg/ml of the following antibodies: rabbit anti-Bak-NT, rabbit anti-Bax-NT (both from Upstate Biotechnology, New York, NY, USA) or 6H2.B4 mouse anti-cytochrome c (Pharmingen, San Diego, CA, USA). After three more washes (each for 10 min) with buffer A, the cells were incubated for 30 min with buffer A containing Cy3-labeled goat anti-mouse or anti-rabbit antibodies (2 μg/ml; Jackson ImmunoResearch). The cells were again washed three times (each for 10 min) with buffer A, air-dried, and mounted with mounting medium (IMMCO Diagnostics, Buffalo, NY, USA). For nuclear staining, cells were subjected to the same procedure except that 1 μg/ml Hoechst dye 33258 (Sigma) was included in the first wash after incubation with Cy3-labeled antibodies. Images were collected on a fluorescence microscope (Olympus, × 100 objective lens) connected to a CCD camera for imaging. Transfected cells expressing GFP-Bcl-xS or GFP and also stained with the corresponding antibodies were counted under the microscope, and the number of GFP-Bcl-xS- or GFP-positive cells that were stained with the antibody was expressed as a percentage of the total number of GFP-Bcl-xS- or GFP-expressing cells. Results of three independent experiments (mean±S.D.) are presented.
Immunoblotting
Total extracts (100 μg protein) from each treatment were separated by 12.5% SDS–PAGE and electroblotted onto supported nitrocellulose. Uniformity of sample loading was verified by Ponceau staining of the blots. Each blot was blocked for 30 min in 10 mM Tris base and 150 mM NaCl, containing 5% fat-free milk, and then incubated for 1 h with mouse anti-FLAG M5 antibody (1 : 1000; Sigma). Goat anti-mouse (1 : 5000; Jackson ImmunoResearch) was used as a second antibody. The blots were developed using an enhanced chemiluminescence kit (Amersham, Arlington Heights, IL, USA).
Assessment of cell survival
Viability of the transfected cells was determined 24, 48, 72, and 96 h after transfection, both by measuring the activity of SEAP in the medium of the transfected cells and by estimating the number of GFP-positive cells visualized by fluorescence microscopy (data not shown). The SEAP activity assay reflects expression of Bcl-xS and SEAP in a pool of cells, whereas estimation of the number of GFP-positive cells reflects Bcl-xS expression on a single-cell basis. SEAP activity was assayed as described elsewhere.52 Briefly, culture medium (200 μl/well in six-well plates) from transfected cells was collected and spun for 2 min at 10 000 × g. The supernatant was incubated at 65°C for 10 min and aliquots (25 μl) from each treatment were then incubated with 200 μl of SEAP buffer (1 M diethanolamine, 0.5 mM MgCl2, 10 mM L-homo-arginine, and 5 mg/ml p-nitrophenyl phosphate; all from Sigma) at 37°C for about 4 h. Assays were performed in triplicate for each treatment. The plates were read on a Micro-ELISA reader at a wavelength of 405 nm. Under these conditions, the OD values (mean±S.E. from at least eight different experiments) obtained for the SEAP activity of different MEFs cotransfected with pcDNA3, pEGFP, and SEAP expression vectors were 0.351±0.045, 0.266±0.053, 0.696±0.093, 0.259±0.05, 0.939±0.095, 0.988± 0.144, 0.549±0.075, and 0.273±0.059 for WT, WT1, WT2, Bax−/−, Bak−/−, DKO, Apaf-1−/−, and caspase-9−/−, respectively.
Colony-formation assay
Subconfluent MEFs grown in six-well plates were cotransfected with the puromycin-resistant plasmid pGKpuro and either pcDNA3 or FLAG-Bcl-xS. After 24 h, the cells were split (1 : 2 and 1 : 20) into 10 cm dishes and grown under conditions of puromycin selection (1–2.5 μg/ml) for 2 weeks. The resulting colonies were fixed with 10% acetic acid in phosphate-buffered saline (PBS), stained with 0.4% crystal violet (Sigma) (dissolved in 10% acetic acid in PBS) solution, and counted. The experiments were carried out in triplicate and repeated at least three times.
Abbreviations
- BH:
-
Bcl-2 homology
- DKO:
-
Bax−/− Bak−/− double knockout
- DMEM:
-
Dulbecco's modified Eagle's medium
- ER:
-
endoplasmic reticulum
- GFP:
-
green fluorescent protein
- OMM:
-
outer mitochondrial membrane
- MEF:
-
mouse embryonic fibroblast
- NT:
-
N terminus
- PBS:
-
phosphate-buffered saline
- SEAP:
-
secreted alkaline phosphatase
- WT:
-
wild type
- TBS:
-
Tris-buffered saline
References
Wang X (2001) The expanding role of mitochondria in apoptosis. Genes Dev. 15: 2922–2933
Lim ML, Lum MG, Hansen TM, Roucou X and Nagley P (2002) On the release of cytochrome c from mitochondria during cell death signaling. J. Biomed. Sci. 9: 488–506
Zheng TS, Hunot S, Kuida K and Flavell RA (1999) Caspase knockouts: matters of life and death. Cell Death Differ. 6: 1043–1053
Gross A, McDonnell JM and Korsmeyer SJ (1999) BCL-2 family members and the mitochondria in apoptosis. Genes Dev. 13: 1899–1911
Borner C (2003) The Bcl-2 protein family: sensors and checkpoints for life-or-death decisions. Mol. Immunol. 39: 615–647
Bouillet P and Strasser A (2002) BH3-only proteins – evolutionarily conserved proapoptotic Bcl-2 family members essential for initiating programmed cell death. J. Cell Sci. 115: 1567–1574
Adams JM and Cory S (2001) Life-or-death decisions by the Bcl-2 protein family. Trends Biochem. Sci. 26: 61–66
Wei MC, Zong WX, Cheng EH, Lindsten T, Panoutsakopoulou V, Ross AJ, Roth KA, MacGregor GR, Thompson CB and Korsmeyer SJ (2001) Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science 292: 727–730
Zong WX, Lindsten T, Ross AJ, MacGregor GR and Thompson CB (2001) BH3-only proteins that bind pro-survival Bcl-2 family members fail to induce apoptosis in the absence of Bax and Bak. Genes Dev. 15: 1481–1486
Cheng EH, Wei MC, Weiler S, Flavell RA, Mak TW, Lindsten T and Korsmeyer SJ (2001) BCL-2, BCL-X(L) sequester BH3 domain-only molecules preventing BAX- and BAK-mediated mitochondrial apoptosis. Mol. Cell 8: 705–711
Sharpe JC, Arnoult D and Youle RJ (2004) Control of mitochondrial permeability by Bcl-2 family members. Biochim. Biophys. Acta 1644: 107–113
Degli Esposti M and Dive C (2003) Mitochondrial membrane permeabilisation by Bax/Bak. Biochem. Biophys. Res. Commun. 304: 455–461
Boise LH, Gonzalez-Garcia M, Postema CE, Ding L, Lindsten T, Turka LA, Mao X, Nunez G and Thompson CB (1993) bcl-x, a bcl-2-related gene that functions as a dominant regulator of apoptotic cell death. Cell 74: 597–608
Dixon EP, Stephenson DT, Clemens JA and Little SP (1997) Bcl-Xshort is elevated following severe global ischemia in rat brains. Brain Res. 776: 222–229
Ferrer I, Lopez E, Blanco R, Rivera R, Ballabriga J, Pozas E and Marti E (1998) Bcl-2, Bax, and Bcl-x expression in the CA1 area of the hippocampus following transient forebrain ischemia in the adult gerbil. Exp. Brain Res. 121: 167–173
Heermeier K, Benedict M, Li M, Furth P, Nunez G and Hennighausen L (1996) Bax and Bcl-xs are induced at the onset of apoptosis in involuting mammary epithelial cells. Mech. Dev. 56: 197–207
Ying WZ, Wang PX and Sanders PW (2000) Induction of apoptosis during development of hypertensive nephrosclerosis. Kidney Int. 58: 2007–2017
Igase M, Okura T, Kitami Y and Hiwada K (1999) Apoptosis and Bcl-xs in the intimal thickening of balloon-injured carotid arteries. Clin. Sci. 96: 605–612
Chandrasekar B, Vemula K, Surabhi RM, Li-Weber M, Owen-Schaub LB, Jensen LE and Mummidi S (2004) Activation of intrinsic and extrinsic proapoptotic signaling pathways in interleukin-18-mediated human cardiac endothelial cell death. J. Biol. Chem. 279: 20221–20233
Yamaguchi H, Inokuchi K and Dan K (2002) The study for loss of bcl-xs expression as a prognostic factor in acute myeloid leukemia. Leuk. Res. 26: 1119–1123
Chang TC, Hung MW, Jiang SY, Chu JT, Chu LL and Tsai LC (1997) Dexamethasone suppresses apoptosis in a human gastric cancer cell line through modulation of bcl-x gene expression. FEBS Lett. 415: 11–15
Clarke MF, Apel IJ, Benedict MA, Eipers PG, Sumantran V, Gonzalez-Garcia M, Doedens M, Fukunaga N, Davidson B, Dick JE, Minn AJ, Boise LH, Thompson CB, Wicha M and Nunez G (1995) A recombinant bcl-x s adenovirus selectively induces apoptosis in cancer cells but not in normal bone marrow cells. Proc. Natl. Acad. Sci. USA 92: 11024–11028
Ealovega MW, McGinnis PK, Sumantran VN, Clarke MF and Wicha MS (1996) bcl-xs gene therapy induces apoptosis of human mammary tumors in nude mice. Cancer Res. 56: 1965–1969
Han JS, Nunez G, Wicha MS and Clarke MF (1998) Targeting cancer cell death with a bcl-XS adenovirus. Springer Semin. Immunopathol. 19: 279–288
Sumantran VN, Lee DS, Woods Ignatoski KM, Ethier SP and Wicha MS (2000) A bcl-xS adenovirus selectively induces apoptosis in transformed cells compared to normal mammary cells. Neoplasia 2: 251–260
Baba M, Iishi H and Tatsuta M (2001) Transfer of bcl-xs plasmid is effective in preventing and inhibiting rat hepatocellular carcinoma induced by N-nitrosomorpholine. Gene Therapy 8: 1149–1156
Lindenboim L, Yuan J and Stein R (2000) Bcl-xS and Bax induce different apoptotic pathways in PC12 cells. Oncogene 19: 1783–1793
Lindenboim L, Borner C and Stein R (2001) Bcl-x(S) can form homodimers and heterodimers and its apoptotic activity requires localization of Bcl-x(S) to the mitochondria and its BH3 and loop domains. Cell Death Differ. 8: 933–942
Braun T, Dar S, Vorobiov D, Lindenboim L, Dascal N and Stein R (2003) Expression of Bcl-x(S) in Xenopus oocytes induces BH3-dependent and caspase-dependent cytochrome c release and apoptosis. Mol. Cancer Res. 1: 186–194
Lindenboim L, Schlipf S, Kaufmann T, Borner C and Stein R (2004) Bcl-x(S) induces an NGF-inhibitable cytochrome c release. Exp. Cell Res. 297: 392–403
Mitra RS, Benedict MA, Qian D, Foreman KE, Ekhterae D, Nickoloff BJ and Nunez G (2001) Killing of sarcoma cells by proapoptotic Bcl-X(S): role of the BH3 domain and regulation by Bcl-X(L). Neoplasia 3: 437–445
Chang BS, Kelekar A, Harris MH, Harlan JE, Fesik SW and Thompson CB (1999) The BH3 domain of Bcl-x(S) is required for inhibition of the antiapoptotic function of Bcl-x(L). Mol. Cell. Biol. 19: 6673–6681
Cartron PF, Moreau C, Oliver L, Mayat E, Meflah K and Vallette FM (2002) Involvement of the N-terminus of Bax in its intracellular localization and function. FEBS Lett. 512: 95–100
Griffiths GJ, Dubrez L, Morgan CP, Jones NA, Whitehouse J, Corfe BM, Dive C and Hickman JA (1999) Cell damage-induced conformational changes of the pro-apoptotic protein Bak in vivo precede the onset of apoptosis. J. Cell Biol. 144: 903–914
Ruffolo SC and Shore GC (2003) BCL-2 selectively interacts with the BID-induced open conformer of BAK, inhibiting BAK auto-oligomerization. J. Biol. Chem. 278: 25039–25045
Desagher S, Osen-Sand A, Nichols A, Eskes R, Montessuit S, Lauper S, Maundrell K, Antonsson B and Martinou JC (1999) Bid-induced conformational change of Bax is responsible for mitochondrial cytochrome c release during apoptosis. J. Cell Biol. 144: 891–901
Kaufmann T, Schlipf S, Sanz J, Neubert K, Stein R and Borner C (2003) Characterization of the signal that directs Bcl-x(L), but not Bcl-2, to the mitochondrial outer membrane. J. Cell Biol. 160: 53–64
Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES and Wang X (1997) Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell 91: 479–489
Wei MC, Lindsten T, Mootha VK, Weiler S, Gross A, Ashiya M, Thompson CB and Korsmeyer SJ (2000) tBID, a membrane-targeted death ligand, oligomerizes BAK to release cytochrome c. Genes Dev. 14: 2060–2071
Zong WX, Li C, Hatzivassiliou G, Lindsten T, Yu QC, Yuan J and Thompson CB (2003) Bax and Bak can localize to the endoplasmic reticulum to initiate apoptosis. J. Cell Biol. 162: 59–69
Scorrano L, Oakes SA, Opferman JT, Cheng EH, Sorcinelli MD, Pozzan T and Korsmeyer SJ (2003) BAX and BAK regulation of endoplasmic reticulum Ca2+: a control point for apoptosis. Science 300: 135–139
Hacki J, Egger L, Monney L, Conus S, Rosse T, Fellay I and Borner C (2000) Apoptotic crosstalk between the endoplasmic reticulum and mitochondria controlled by Bcl-2. Oncogene 19: 2286–2295
Donovan M and Cotter TG (2004) Control of mitochondrial integrity by Bcl-2 family members and caspase-independent cell death. Biochim. Biophys. Acta 1644: 133–147
Putcha GV, Moulder KL, Golden JP, Bouillet P, Adams JA, Strasser A and Johnson EM (2001) Induction of BIM, a proapoptotic BH3-only BCL-2 family member, is critical for neuronal apoptosis. Neuron 29: 615–628
Gillissen B, Essmann F, Graupner V, Starck L, Radetzki S, Dorken B, Schulze-Osthoff K and Daniel PT (2003) Induction of cell death by the BH3-only Bcl-2 homolog Nbk/Bik is mediated by an entirely Bax-dependent mitochondrial pathway. EMBO J. 22: 3580–3590
Scorrano L and Korsmeyer SJ (2003) Mechanisms of cytochrome c release by proapoptotic BCL-2 family members. Biochem. Biophys. Res. Commun. 304: 437–444
Cheng EH, Sheiko TV, Fisher JK, Craigen WJ and Korsmeyer SJ (2003) VDAC2 inhibits BAK activation and mitochondrial apoptosis. Science 301: 513–517
Tao W, Kurschner C and Morgan JI (1997) Modulation of cell death in yeast by the Bcl-2 family of proteins. J. Biol. Chem. 272: 15547–15552
Griffiths GJ, Corfe BM, Savory P, Leech S, Esposti MD, Hickman JA and Dive C (2001) Cellular damage signals promote sequential changes at the N-terminus and BH-1 domain of the pro-apoptotic protein Bak. Oncogene 20: 7668–7676
Yoshida H, Kong YY, Yoshida R, Elia AJ, Hakem A, Hakem R, Penninger JM and Mak TW (1998) Apaf1 is required for mitochondrial pathways of apoptosis and brain development. Cell 94: 739–750
Todaro GJ and Green H (1963) Quantitative studies of the growth of mouse embryo cells in culture and their development into established lines. J. Cell Biol. 17: 299–313
Berger J, Hauber J, Hauber R, Geiger R and Cullen BR (1988) Secreted placental alkaline phosphatase: a powerful new quantitative indicator of gene expression in eukaryotic cells. Gene 66: 1–10
Acknowledgements
We are grateful to Ms. Shirley Smith for excellent editorial assistance. We thank Drs. S Korsmeyer, A Strasser, and T Mak for providing us with the different MEFs. This work was supported by the German–Israeli Foundation (to RS and CB) and Deutsche Forschungsgemeinschaft (BO-1933) (to CB).
Author information
Authors and Affiliations
Corresponding author
Additional information
Edited by D Vaux
Rights and permissions
About this article
Cite this article
Lindenboim, L., Kringel, S., Braun, T. et al. Bak but not Bax is essential for Bcl-xS-induced apoptosis. Cell Death Differ 12, 713–723 (2005). https://doi.org/10.1038/sj.cdd.4401638
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.cdd.4401638
Keywords
This article is cited by
-
LINC complex protein nesprin-2 has pro-apoptotic activity via Bcl-2 family proteins
Cell Death Discovery (2024)
-
Apoptosis in Alzheimer’s disease: insight into the signaling pathways and therapeutic avenues
Apoptosis (2023)
-
Apoptotic stress induces Bax-dependent, caspase-independent redistribution of LINC complex nesprins
Cell Death Discovery (2020)
-
BCL-xL/BCL2L1 is a critical anti-apoptotic protein that promotes the survival of differentiating pancreatic cells from human pluripotent stem cells
Cell Death & Disease (2020)
-
BCL-2 family isoforms in apoptosis and cancer
Cell Death & Disease (2019)
