
Abstract
Chronic hepatitis C progresses to cirrhosis within 20 years in an estimated 20–30% of patients, while running a relatively uneventful course in most others. Certain HCV proteins, such as core and NS5A, can induce derangement of lipid metabolism or alter signal transduction of infected hepatocytes which leads to the production of reactive oxygen radicals and profibrogenic mediators, in particular TGF-β1. TGF-β1 is the strongest known inducer of fibrogenesis in the effector cells of hepatic fibrosis, i.e. activated hepatic stellate cells and myofibroblasts. However, fibrogenesis proceeds only when additional profibrogenic stimuli are present, e.g. alcohol exposure, metabolic disorders such as non-alcoholic steatohepatitis, or coinfections with HIV or Schistosoma mansoni that skew the immune response towards a Th2 T cell reaction. Furthermore, profibrogenic polymorphisms in genes that are relevant during fibrogenesis have been disclosed. This knowledge will make it possible to identify those patients who are most likely to progress and who need antiviral or antifibrotic therapies most urgently. However, even the best available treatment, the combination of pegylated interferon and ribavirin, which is costly and fraught with side effects, eradicates HCV in only 50% of patients. While the suggestive antifibrotic effect of interferons (IF-γ>α,β), irrespective of viral elimination, has to be proven in randomised prospective studies, additional, well tolerated and cost-effective antifibrotic therapies have to be developed. The combination of cytokine strategies, e.g. inhibition of the key profibrogenic mediator TGF-β, with other potential antifibrotic agents appears promising. Such adjunctive agents could be silymarin, sho-saiko-to, halofuginone, phosphodiesterase inhibitors, and endothelin-A-receptor or angiotensin antagonists. Furthermore, drug targeting to the fibrogenic effector cells appears feasible. Together with the evolving validation of serological markers of hepatic fibrogenesis and fibrolysis an effective and individualised treatment of liver fibrosis is anticipated.
Similar content being viewed by others

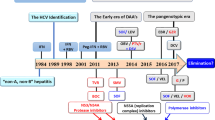

Natural evolution of hepatitis C infection
Acute infection with the hepatitis C virus is uneventful in most of the cases, but runs a chronic course in an estimated 70% of patients. Because of the paucity of early symptoms, most infections are either diagnosed by chance due to transaminase elevations or in an advanced disease stage, when complications have occurred. Development of cirrhosis is the main determinant of morbidity and mortality, presenting either in a compensated state with only minor abnormalities of liver function or with fully decompensated liver disease which usually portends a dire prognosis. Furthermore, compensated as well as decompensated cirrhosis is a crucial precondition for the development of hepatocellular carcinoma (HCC), with an estimated yearly incidence of 2–5% in cirrhotics with hepatitis C. Consequently, only those patients whose HCV infection has led to cirrhosis or tends to progress to cirrhosis within a fairly short time period pose a major health problem, whereas patients who do not progress or who progress slowly usually do not require antiviral or antifibrotic treatment.
The natural evolution of chronic hepatitis C has been analyzed in a large retrospective study from France.1 Here, roughly one third of patients progressed to cirrhosis in approximately 20 years, one third within approximately 50 years, and one third did not show any progression (Figure 1). Apart from duration of infection, factors associated with advanced fibrosis were male gender, age >40 years, older age at infection, genotype 1 and alcohol abuse. However, the fraction of HCV patients without progression may be higher in nonselected patients, since the French study mainly considered cases that came to attention due to clinical or laboratory abnormalities. Thus the expected number of HCV infections without such abnormalities, e.g. including cases with persistently normal transaminases which predicts a less severe course of the disease, is higher.2,3

Natural course of hepatitis C and sustained response (SR) to antiviral therapy. Numbers as to the natural course of the disease are derived from retrospective studies; GT, genotype; NASH, non-alcoholic steatohepatitis
Antiviral treatment
Antiviral treatment is given to patients who fulfil the criteria of chronic hepatitis (>6 months duration), with elevated transaminases and at least some degree of histological inflammation or fibrosis. In hepatitis C, transaminases and histology do predict insufficiently whose disease will progress in a short time. But additional predictive parameters of disease progression are being validated the clinical application of which is pending.
Using the best available therapy, i.e. pegylated interferon alfa and optimal doses of ribavirin, virus negativity 6 months after discontinuation of treatment (sustained response) can be achieved in 40–50% and 80% of patients with genotype 1 and 2/3, respectively.4,5 Retrospective analysis suggests that viral elimination stops disease progression and may even induce reversion of fibrosis.6 Despite these promising results, at least 50% of patients with genotype 1 which is the most frequent genotype in the Western world (60–90% of those infected), or with genotype 4 (which is prominent in Egypt) do not respond to this optimised antiviral therapy, let alone the patients who have contraindications to interferon or ribavirin. Moreover, the costs of interferon and ribavirin are out of reach for many health care systems and this combination has significant side effects. Therefore, reasonable pharmacological alternatives that retard fibrosis progression are needed.
Mechanisms of hepatic fibrogenesis
Chronic liver diseases frequently lead to scarring (cirrhosis) which is often accompanied by progressive loss of liver function despite the use of immunosuppressive, antiviral or antiinflammatory agents.
Fibrosis results from excessive accumulation of extracellular matrix (ECM). The collagens are the most important molecular targets, since (1) they represent the major matrix proteins, (2) they form important mechanical scaffolds and (3) their proteolysis by specific proteases appears to be rate-limiting for ECM removal. The fibril forming interstitial collagens type I and III, and the sheet-forming basement membrane collagen type IV are the most abundant ECM components in liver. In cirrhosis their content increases up to 10-fold.7 A variety of adverse stimuli such as toxins, viruses, bile stasis, hypoxia can trigger fibrogenesis, i.e., the excess synthesis and deposition of ECM, usually by activation of cytokine release, or simply by mechanical stress. In the acute phase of liver disease fibrogenesis is balanced by fibrolysis, i.e., the removal of excess ECM by proteolytic enzymes, the most important of which are the matrix metalloproteinases (MMPs). MMP-1, -2, -3, -8, -9,-12, -13 and -14 are expressed in human liver.8 With repeated injury of sufficient severity, fibrogenesis prevails over fibrolysis, resulting in excess ECM deposition, i.e. fibrosis. Fibrogenesis is characterised by an upregulation of ECM synthesis, a downregulation of MMP secretion and activity, and by an increase of the physiological inhibitors of the MMPs, the tissue inhibitors of MMPs (TIMPs). Among the four known TIMPs, the universal MMP-inhibitor TIMP-1 is most important.9 However, an increase of certain MMPs may also be detrimental. Thus activation of MMPs at the wrong place and time can lead to removal of the regular, differentiation-inducing ECM, such as basement membranes, with subsequent unfavourable tissue remodelling, architectural distortion and a fibrogenic response. An example is MMP-2 which mainly degrades basement membrane collagen and denatured collagens and which is upregulated during fibrogenesis. Collagens, MMPs and TIMPs are mainly produced by myofibroblastic cells (MF) which either derive from activated hepatic stellate cells (HSC) or from activated (portal and perivascular) fibroblasts10,11 (Figure 2). Activated liver macrophages, i.e. Kupffer cells, or proliferating bile ductular epithelia, but also endothelia, other mononuclear cells and myofibroblasts themselves are sources of fibrogenic cytokines and growth factors that can stimulate HSC and perivascular fibroblasts to become MF. The prominent profibrogenic cytokine is transforming growth factor β1 (TGF-β1) which is released from almost any cell during inflammation, tissue regeneration and fibrogenesis. Apart from immunosuppressive and, in most cell types, antiproliferative effects TGF-β1 strongly upregulates production and deposition of the major ECM molecules.11,12,13 Therefore, TGF-β1 as well as activated HSC and MF are the prime targets for antifibrotic therapies (Figure 2).

Initiation and maintenance of fibrogenesis. With continuous injury, primarily to hepatocytes or bile duct epithelia, and/or mechanical stress the normally quiescent hepatic stellate cells and portal fibroblasts undergo activation and transdifferentiation to myofibroblasts. These myofibroblasts produce excessive amounts of collagens, downregulate matrix metalloproteinases (MMPs) and show an enhanced expression of the physiological inhibitors of the MMPs (TIMP-1 and -2). TIMP-1 can also promote myofibroblast proliferation and inhibit their apoptosis
Genetic predisposition for hepatic fibrosis
Genetic polymorphisms may explain the widely differing individual rates of fibrosis progression in chronic hepatitis C. Several polymorphisms of genes that are involved in inflammation, immune regulation and regeneration have been implicated in autoimmune diseases, and some of these are candidates for the regulation of hepatic fibrogenesis (Table 1). While there is some minor contribution by IL-10 and TNF-α gene polymorphisms, a recent study from Australia showed that a promoter polymorphism at position −6 of the angiotensin gene and a mutation causing a proline to arginine substitution at amino acid position 25 of the TGFβ1 precursor confer susceptibility to accelerated fibrogenesis.14 Thus HCV patients with both polymorphisms exhibited fibrosis stage 2.5 (with four being cirrhosis) compared to 0.5 for patients without either of these polymorphisms. Furthermore, the HLA-DRB1*11 allele appears to protect HCV patients from progression to cirrhosis after liver transplantation.15
Contribution of a second hit
There is increasing evidence that severity and progression of fibrosis in chronic liver diseases is not only caused by repetitive exposure to a single detrimental external or internal factor, but by additional events that exacerbate the condition. This has been shown for the combination of hemochromatosis with alcohol consumption or with chronic viral hepatitis,16,17,18 of alcoholic liver disease with chronic viral hepatitis,19 and for the contribution by non-alcoholic steatohepatitis, a disorder related to the metabolic syndrome.19,20,21 Thus even the consumption of as little as 10 g of alcohol per day was associated with a significant effect on histological fibrosis progression in HCV patients.19 In contrast, the natural course of two large cohorts of young women from Ireland and Germany who were infected during Rhesus immunoprophylaxis more than 20 years ago, has been uneventful, with less than 2% developing cirrhosis and an estimated mean progression to cirrhosis in more than 60 years.22,23 Similarly, among asymptomatic HCV-infected blood donors, none developed cirrhosis 19–24 years after exposure.24
The fibrogenic immune response
Immune-mediated damage is linked to fibrogenesis, but the character of the immune response determines fibrosis progression. Contrary to general thinking, fibrosis appears not to be the logical consequence of significant macroscopic inflammation as reflected by the mononuclear infiltrate, but rather by the associated intrinsic or extrinsic immunosuppression. Here cytokines (growth factors) play a prominent role. They can be subdivided into a fibrogenic, antifibrogenic and neutral group. The latter has by definition no effect on the fibrogenic effector cells, i.e. activated HSC and MF. As a rule of thumb certain Th1 cytokines, i.e. those that stimulate cellular immune responses, rather trigger matrix dissolution, i.e., fibrolysis, whereas Th2 cytokines, which stimulate the humoral immune response and can suppress Th1 T cells, promote fibrogenesis. Examples are the interferons (Th1) on one hand,6,25,26 and transforming growth factor β1 (TGF-β1),11,12,13 interleukin-4 (IL-4) and most likely IL-10 on the other hand.27 Evidence comes from patients coinfected with both HIV (Th2) and HCV (Th1 in acute, Th2 in chronic infection) who progress more quickly to cirrhosis than patients infected with HCV alone.28 Similarly, accelerated fibrosis progression is found in HCV patients coinfected with Schistosoma mansoni which triggers a Th2 cytokine profile.29
Are there fibrogenic HCV proteins?
A major impediment to development of specific antiviral drugs has been the lack of a highly replicative and low cost in vitro or in vivo model of HCV infection. This may change after the recent generation of human hepatoma cell lines that persistently replicate HCV30 and of a mouse model that harbours HCV-replicating human hepatocytes.31 Several studies with HCV-transfected, preferably hepatocytic cell lines and HCV-transgenic mouse models have emerged in recent years32 that enable some conclusions to be drawn as to possible direct profibrogenic and procarcinogenic mechanisms of certain HCV-proteins, irrespective of the host's immune response. However, only some transgenic mouse strains develop liver damage, pointing to additional, genetic predispositions. Furthermore, numerous studies used a highly expressed single gene or an incomplete set of HCV-genes, a setting that does not mimic human infection which is usually characterised by moderate viral replication in liver. Finally, expression of a limited set of genes ignores the interactive potential of HCV proteins with each other.33
Overexpression of the cytoplasmatically located HCV core has been analysed in detail. Sensitive C57BL/6 mice with the core transgene driven by the albumin promoter develop hepatic lipid accumulation (steatosis) after 6 months and hepatocellular carcinoma (HCC) after 16 months.34,35 In cell culture the core protein has transforming potential.36 It can interact with the intracytoplasmic TNF-α type 1 and the lymphotoxin β receptors, enhancing their pro-apoptotic signal transduction,37,38 and activate the tumour suppressor p53.39 In addition, HCV core can repress the cell cycle regulator p21WAF40 and via inhibition of the p38 mitogen activated kinase pathway promote Fas-induced apoptosis.41 Taken together, these data rather suggest a pro-apoptotic effect of core protein in hepatocytes, favouring the elimination of infected and potentially transformed cells. On the other hand, anti-apoptotic Bcl-xl can be upregulated.42 While the jury is open as to a predominatly anti- or proapoptic effect of core protein, the data regarding lipid accumulation in core-transfected hepatocytes, one of the second hits that favour hepatic fibrogenesis,20 are more homogeneous. Thus it interacts with apolipoprotein II and reduces microsomal triglyceride transfer protein in vivo, inhibiting assembly and secretion of regular VLDL particles.43 In transfected hepatoma cells core activates the retinoid X receptor alpha which dimerises with the peroxisome proliferator activated receptor alpha to upregulate genes of lipid metabolism like cellular retinol binding protein II and acetyl CoA-reductase.44 Notably, by interaction with mitochondria core protein induces reactive oxygen intermediates and thus oxidative stress which can induce steatohepatitis.45,46
The non-structural protein NS5A has also been implicated in HCV pathogenicity. It can compromise the antiviral and hypothetically antifibrotic effect of interferon, obviously either by repression of protein kinase R or alternative pathways47,48. NS5A enhances the acute phase response via activation of NF-kappa B and STAT-3 and, as core, causes oxidative stress.49 In addition, NS5A has been implicated in favouring cell cycle progression to the G2/M by binding to the cyclin dependent kinase 1 (cdk1)/cdk2-complex.50
Transgenic mice that express very low levels of the complete reading frame of the HCV polyprotein developed significantly more spontaneous HCCs after 13 months compared to their nontransgenic controls (5/38 vs 0/16), while transgenic mice expressing significant levels of the HCV structural proteins (Core, E1 and E2) only developed HCC in a single case (1/43 vs 0/35).51 While steatosis developed in all animals, inflammation, apoptosis and histochemical fibrosis were not enhanced in both models, and increased hepatocellular proliferation was only found in the transgenic mice harbouring the complete HCV genome. Matrix gene expression remains to be investigated in these models which should also be challenged, to test how far a second hit can promote both fibrosis and hepatocarcinogenesis. Potential profibrogenic actions of HCV-proteins are illustrated in Figure 3.

HCV proteins as triggers of inflammation, apoptosis and fibrogenesis. Core, NS3 and NS5A have been studied most intensively. All three can interact with mitochondria and increase the formation of reactive oxygen species (ROS). Core and NS5A associate with the membranes of the endoplasmatic reticulum and golgi apparatus. In particular, they can enhance intracellular lipid accumulation via interactions with apolipoprotein A1 (apoA1) or A2 (apoA2) which cause inhibition of lipid transfer protein or defective synthesis and transport of very low density lipoprotein (VLDL). Core has also been shown to activate the retinoid X receptor (RXRα) which by dimerisation with the peroxisome proliferator activated receptor alpha (PPAR-α) induces proinflammatory NFkB, cytochrome P450A4 (CYPA4) (which combined with steatosis favours lipid peroxidation and thus ROS formation) and lipid degradation via enhanced β-oxidation (leading to reduction of VLDL production). Taken together, the HCV proteins appear to induce both lipid accumulation and degradation, with consequent derangement of lipid compartmentalisation and metabolism, favouring ROS production. ROS lead to induction of TGF-β, the strongest known promoter of fibrogenesis. Core has also been shown to trigger apoptosis by interaction with TNF receptors and via downregulation of p21 and p38, and upregulation of p53, while NS5A can induce inflammation via NFkB and stat-3, and cell cycle progression via activation of the cyclin dependent kinase cdk1/cdk2 complex
Serum markers of liver fibrosis
An index that uses α2-macroglobulin, γ-globulin, haptoglobin, γ-GT and bilirubin could predict the absence or the presence of significant fibrosis/cirrhosis (metavir stage 2–4) in 46% of patients with chronic hepatitis C.52 While 54% of the patients could not be allocated, this index will not be suited to detect day-to-day changes in the active processes of fibrogenesis or fibrolysis. Classical serum fibrosis markers are matrix constituents that are released into the circulation during matrix remodelling. The majority of these markers appear to reflect fibrogenesis rather than fibrolysis53 (Figure 4). These surrogate markers may open the possibility to assess the future evolution of fibrosis and the effect of potential antifibrotic treatment in an individual patient and on a frequent basis. Several studies to validate these markers in large prospective follow-up studies of patients with liver diseases are currently under way. The availability of serological fibrosis markers is central to the rapid clinical development and validation of antifibrotic therapies.

Serum markers of fibrogenesis and fibrolysis. Procollagen precursors released by fibrogenic cells are processed by procollagen peptidases. Removal of the bulky propeptides allows the formation of collagen fibrils in the extracellular space. Thus circulating propeptide levels should reflect de novo synthesis and deposition of collagen, i.e. fibrogenesis. On the other hand, action of matrix metalloproteinases (MMPs) is expected to generate fragments of already deposited matrix proteins the levels of which should reflect matrix dissolution, i.e. fibrolysis. The diagnostic and predictive value of these markers is currently evaluated
Antifibrotic drug development
A major obstacle to antifibrotic drug development is the slow evolution of significant fibrosis which takes years or even decades in man. Sequential biopsy with semiquantitative and preferably quantitative assessment of fibrous tissue remains the gold standard to monitor fibrosis progression, but sampling error remains a problem, since due to large regenerative nodules that consist mainly of hepatocytes up to 25% of patients with advanced fibrosis or cirrhosis may be wrongly categorised as only slightly fibrotic.54 Consequently, prospective studies in patients have to be large and testing of the large spectrum of potential antifibrotic agents is impossible. First proof of efficacy has to come from cell culture data that show inhibition of proliferation, induction of apoptosis and/or downregulation of collagen production in the key fibrogenic liver cells, i.e. activated HSC and MF. This has to be followed by suitable animal models of hepatic fibrosis to show the antifibrotic effect in vivo in the absence of general toxicity. Rat models are preferable, since significant fibrosis can be produced within 3–10 weeks and, most importantly, total liver collagen, the gold standard for fibrosis, can be determined easily in a representative tissue sample using biochemical methods. However, the in vivo models must include a sizeable number of animals per treatment group (n=10–20) and should be devoid of major hepatocyte necrosis. This is important, since drugs with anti-inflammatory, anti-necrotic or radical scavenging properties can prevent necrosis and collapse, as is characteristic of the models induced by carbon tetrachloride, dimethylnitrosamine or galactosamine, but are not truly antifibrotic. Thus fibrosis should evolve chronically and reproducibly, with no or little inflammation and necrosis, as in biliary cirrhosis due to bile duct occlusion. In addition, models of fibrosis reversion, e.g. after induction by carbon tetrachloride or thioacetamide, are alternatives to predict a true antifibrotic drug effect. Many reports on so-called antifibrotic agents do not fulfil the above-mentioned criteria, and the following examples will refer mainly to those studies that provide sufficient in vivo evidence for an antifibrotic effect. Importantly, on the long term a decrease of fibrosis should be followed by an improvement of portal hypertension and liver function.
Cytokine strategies to inhibit hepatic fibrosis
Antifibrotic cytokines
While immunosuppressive or Th2 cytokines are usually profibrogenic,27,28,29 most Th1 cytokines, especially the antiviral interferons are antifibrogenic. This effect is linked, at least in part, to the activation of stat-1 signalling pathways (Figure 5). In cell culture interferon-α (IF-α) blocks activation, proliferation and collagen synthesis of HSC and MF.25 Retrospective analyses and one small prospective study in patients with chronic hepatitis C suggest that IF-α therapy can prevent fibrosis progression, even in nonresponders to antiviral therapy.1,2,6,26 The effect was dependent on IF-α dose duration, and most pronounced in sustained responders. A recent analysis of four large international studies even found reversion of cirrhosis in 75 out of 153 patients.6 However, there might be a problem of sampling error and the number of initially noncirrhotic patients who had cirrhosis on the second biopsy is not stated. Several large prospective studies have been initiated (HALT-C, EPIC-3 and COPILOT, PROFI-C) (Table 2) that try to answer the question of an antifibrotic effect of longterm IF-α (up to 4 years) in nonresponders to IF-α/ribavirin combination therapy by using conventional follow-up histology. In PROFI-C nonresponders to combination therapy receive retarded (pegylated) IF-α at 100 or 25 g per week combined with high-dose (840 mg/day) silymarin or placebo for 2 years. Fibrosis progression is assessed by collagen morphometry of pre- and post-treatment biopsies and by sequential measurement of a spectrum of serum fibrosis markers.

Effects of interferon-α and -β on fibroblast signaling and gene transcription. Interferon-α and -β activate the same dimeric receptor which leads to recruitment of the intracellular tyrosine kinases jak-1 and tyk-2. This receptor complex catalyses the assembly of stat-1, stat-2 and p48 to a trimeric complex that directs transcription of genes involved in the known interferon-induced effects including inhibition of proliferation and collagen production
Antagonising profibrogenic cytokines
TGF-β1 is considered the most potent fibrogenic cytokine and its inhibition therefore appears attractive.11,12,13 Soluble TGF-β1 decoy receptors or adenoviral constructs that block TGF-β1 signalling have been developed that show antifibrotic efficacy in vitro and in vivo13,55,56,57 (Figure 6). It appears that an approach targeting activated HSC and MF is necessary, since TGF-β receptors are expressed on most cell types and systemic inhibition that reaches sufficient levels to block hepatic fibrogenesis may trigger autoimmune diseases and cellular de-differentiation (Figure 7). Connective tissue growth factor (CTGF),58 a fibrogenic cytokine that is triggered by TGF-β, is expressed mainly in hepatic mesenchymal cells, but also in proliferating bile duct epithelia. Its inhibition may render a more specific antifibrotic strategy. Reports on the antifibrotic activity of hepatocyte growth factor (HGF)59 have to be interpreted with care, since HGF rather causes hypertrophy and hyperplasia of (liver) epithelia, thus reducing the relative and not the absolute collagen content in the organ, with an inherent risk of promoting epithelial malignancy. IL-10, an immunosuppressive cytokine, was reported to reduce liver fibrosis in a study of 22 patients with chronic hepatitis C who were treated for only 12 weeks. However, apart from IL-10 being a Th2-like cytokine for which a profibrogenic effect may be anticipated, there is an inherent problem of sampling error in a small study like this.27

Anti-TGF-β1 strategies to inhibit fibrogenesis. Strategies have been developed that neutralize TGF-β1, e.g. by blockage or inactivation of its signaling receptors (T-βRI or T-βRII), the major signal transducers of the TGF-β pathway, such as smad2 and smad3, or SARA (smad anchor for receptor activation), a protein that allows docking of smad2 to the activated T-βRII. Alternatively, the inhibitory smad7 which is induced as an endogenous feed-back inhibitor late after activation of profibrogenic genes can be delivered by gene transfer

Biological actions of TGF-β1. Apart from initiating a profibrogenic response in mesenchymal cells, TGF-β1 is an essential mediator controlling Th1 immune activation or cellular dedifferentiation. Its long-term untargeted inhibition may trigger autoimmunity and neoplasia
Other antifibrotic agents and combination therapies
Plant-derived drugs
Several promising drugs derive from plants. Silymarin from the milk thistle contains three prominent flavonoids, with silibinin representing up to 60% of the dried extract. Silibinin was shown to stimulate hepatocyte RNA synthesis, to act as a radical-scavenger and hepatoprotectant, and to suppress HSC proliferation and collagen synthesis in vitro. Importantly, it reduced hepatic collagen accumulation in rat biliary fibrosis secondary to bile duct occlusion, a model which leads to a 10–12-fold hepatic collagen accumulation after 6 weeks, by 30–40%, even when treatment was started in an advanced stage of fibrosis.60 The major alkaloid baicalein from the traditional Chinese/Japanese plant extract Sho-saiko-to that displays a structure similar to silibinin has radical-scavenging but also antifibrotic properties in activated HSC in vitro and in rat porcine serum-induced fibrosis in vivo.61 Halofuginone, a semisynthetic alkaloid-derivative from the antimalarial plant Dichroa febrifuga, was shown to normalise hepatic collagen content in a fibrosis reversion model after induction of hepatic fibrosis by thioacetamide.62 These drugs appear to mainly act as anti-oxidants, though probably with different pharmacokinetics, pharmacodynamics and cellular specificities. Intracellular oxidative stress is a relevant contributor to fibrogenesis, and recent studies have shown the induction of profibrogenic TGF-β1 by peroxide radicals,63,64 providing a rationale for the use of intracellular anti-oxidants as adjunctive antifibrotic agents (Figure 8).

TGF-β1, oxidative stress and antioxidants. Oxidative stress and the generation of hydrogen peroxide activate the TGF-β1 pathway and vice versa. Certain antioxidants may therefore exert antifibrotic effects. Drugs that act mainly extracellularly, like vitamin E, are expected to be less effective. NAc, N-acetyl cysteine; SAMe, S-adenosyl-methionine
Modulators of fibrogenic signal transduction
In vitro the phosphodiesterase inhibitor and cytokine antagonist pentoxifylline suppresses proliferation and collagen production by skin fibroblasts and HSC,65 while in rat biliary fibrosis, oral pentoxifylline reduced hepatic collagen accumulation by only 20%. The drug induced a hitherto unmatched eightfold downregulation of hepatic procollagen I mRNA, the product of activated HSC and MF, but this was counterbalanced by a twofold increase of hepatic TIMP-1 mRNA expression, with pentoxifylline apparently stimulating bile duct epithelia and hepatocytes to express TIMP-1.66 Better targeting of pentoxifylline to HSC and MF may prevent upregulation of the profibrogenic TIMP-1 without compromising the downregulatory effect on procollagen I expression.
Antagonizing vasoactive mediators
Oral endothelin A receptor (ETAR) antagonists are attractive, since the ETAR mediates HSC/MF contraction, proliferation and possibly also collagen synthesis, whereas the ETBR induces MF relaxation and inhibition of proliferation. In rat biliary fibrosis the oral ETAR antagonist LU135252 reduced hepatic collagen accumulation by up to 60% when given over the full 6 weeks of the experiment, being still effective when treatment was begun after week 3, a time point with an already fourfold increased liver collagen.67 Angiotensin 1 receptor antagonists or angiotensin converting enzyme inhibitors can retard liver fibrosis in suitable rat models.68,69 However, since the doses applied are up to 100-fold above the doses given in human hypertension, their antifibrotic effect in man remains controversial.
Modulation of growth factor bioactivity by matrix-derived peptides
Certain growth factors and cytokines can bind to matrix molecules. Basic fibroblast growth factor, some hematopoietic growth factors, platelet-derived growth factor (PDGF), HGF, keratinocyte growth factor (KGF) and others are immobilised and protected from proteolytic degradation by heparan sulphates. More importantly, the epithelial mitogens HGF and KGF, the profibrogenic mitogens PDGF, oncostatin M, and the pro-inflammatory cytokine IL-2 bind to the abundant collagens.70,71,72 This binding is reversible and retains the factors in a biologically active form in the fibrotic matrix. The collagenous consensus peptide (Gly-Pro-Hyp)n can liberate the collagen-bound factors from the matrix.72 It remains to be shown how far application of this peptide can perturb matrix metabolism in fibrogenesis.
Reversion of the fibrogenic phenotype
A promising target is the induction of stress relaxation of fibrogenic cells, a matrix (integrin) receptor-mediated process that is associated with a decrease in collagen synthesis and an increase in collagenase activity.73,74 Stress relaxation occurs once mesenchymal cells are placed from a stressed, two-dimensional environment (mimicking a situation of wounding) into a relaxed, three-dimensional context. Thus, the receptors for platelet-derived growth factor,75 endothelin-1 (via the ETAR) or the pericellular collagen VI76 transmit stress signals that trigger proliferation and ECM synthesis in activated HSC and MF. Apart from the possibility to directly block these receptors, coupling receptor-ligands to a drug carrier allows a highly specific targeting to the activated fibrogenic cells in the liver. This has been shown both in vitro and in vivo with cyclic peptides recognising the receptors for PDGF, mannose-6-phosphate and collagen VI. More than 40% of the i.v. injected collagen VI receptor recognising construct was found bound to activated HSC of fibrotic rat livers, and in vitro data demonstrated its efficient internalisation.77 Therefore, such carriers to receptors that are highly upregulated on activated HSC and MF are promising vehicles for targeted delivery of various antifibrotic agents (Figure 9).

Targeting of activation-dependent receptors. Receptors for collagen VI (CVI) or PDGF75,76,77 and other profibrogenic mediators are predominantly upregulated on activated hepatic stellate cells/myofibroblasts. Their respective ligands are in part produced in an autocrine and paracrine manner by the activated cells themselves. These receptors can either be blocked directly by use of antagonistic small peptides or peptide analogues, or serve as ideal targets to deliver potential antifibrotic agents exclusively to the fibrogenic focus. Antifibrotic agents can be coupled to these receptor-recognizing molecules, leading to highly efficient delivery to the fibrogenic cells, followed by internalisation with release of the antifibrotic priniciple. CTGF, connective tissue growth factor; antisense, antisense constructs directed to profibrogenic mRNAs
Combination therapy for hepatic fibrosis
As in cancer therapy combination of several drugs that show different actions by either blocking fibrogenesis, stimulating fibrolysis, by inducing myofibroblast apoptosis, or reversion to a fibrolytic phenotype is most promising. This will make it possible to use lower, non-toxic amounts of single agents for a treatment that will have to be given for years or even life-long. Table 3 lists additional drugs that may be useful in such combinations.
Abbreviations
- CTGF:
-
connective tissue growth factor
- ET:
-
endothelin
- ETAR:
-
endothelin A receptor
- HGF:
-
hepatocyte growth factor
- HCC:
-
hepatocellular carcinoma
- MF:
-
myofibroblastic cells
- MMP:
-
matrix metalloproteinase
- MF:
-
myofibroblast
- PDGF:
-
platelet derived growth factor
- HSC:
-
hepatic stellate cell
- TGF:
-
transforming growth factor
- TIMP:
-
tissue inhibitor of metalloproteinases
References
Poynard T, Ratziu V, Charlotte F, Goodman Z, McHutchison J & Albrecht J (2001) Rates and risk factors of liver fibrosis progression in patients with chronic hepatitis C. J. Hepatol. 34: 730–739.
Manns MP, McHutchison JG, Gordon SC, Rustgi VK, Shiffman M, Reindollar R, Goodman ZD, Koury K, Ling M & Albrecht JK (2001) Peginterferon alfa-2b plus ribavirin compared with interferon alfa-2b plus ribavirin for initial treatment of chronic hepatitis C: a randomised trial. Lancet 358: 958–965.
Mathurin P, Moussalli J, Cadranel JF, Thibault V, Charlotte F, Dumouchel P, Cazier A, Huraux JM, Devergie B, Vidaud M, Opolon P & Poynard T (1998) Slow progression rate of fibrosis in hepatitis C virus patients with persistently normal alanine transaminase activity. Hepatology 27: 868–872.
Alter MJ, Kruszon-Moran D, Nainan OV, McQuillan GM, Gao F, Moyer LA, Kaslow RA & Margolis HS (1999) The prevalence of hepatitis C virus infection in the United States, 1988 through 1994. N. Engl. J. Med. 341: 556–562.
Hadziyannis SJ, Cheinquer H, Morgan T, Diago M, Jensen DM, Sette, Jr H, Ramadori G, Bodenheimer HC & Marcellin P et al (2002) Peginterferon alfa-2A (40 KD) (PEGASYS) in combination with ribavirin (RBV): efficacy and safety results from a phase III, randomized, double-blind, multicentre study examining effect of duration of treatment and RBV dose. J. Hepatol. 36: (Suppl 1) 3
Poynard T, McHutchison J, Manns M, Trepo C, Lindsay K, Goodman Z, Ling MH & Albrecht J (2002) Impact of pegylated interferon alfa-2b and ribavirin on liver fibrosis in patients with chronic hepatitis C. Gastroenterology 122: 1303–1313.
Schuppan D, Ruehl M, Somasundaram R & Hahn EG (2001) Matrix as a modulator of hepatic fibrogenesis. Sem. Liver Dis. 21: 351–372.
Benyon RC & Arthur MJ (2001) Extracellular matrix degradation and the role of hepatic stellate cells. Semin. Liver Dis. 21: 373–384.
Iredale JP Tissue inhibitors of metalloproteinases in liver fibrosis. (2001) Int. J. Biochem. Cell Biol. 29: 43–54.
Knittel T, Kobold D, Saile B, Grundmann A, Neubauer K, Piscaglia F & Ramadori G (1999) Rat liver myofibroblasts and hepatic stellate cells: different cell populations of the fibroblast lineage with fibrogenic potential. Gastroenterology 117: 1205–1221.
Friedman SL (2000) Molecular regulation of hepatic fibrosis, an integrated cellular response to tissue injury. J. Biol. Chem. 275: 2247–2250.
Bissell DM, Roulot D & George J (2001) Transforming growth factor β and the liver. Hepatology 34: 859–867.
Gressner AM, Weiskirchen R, Breitkopf K & Dooley S (2002) Roles of TGF-beta in hepatic fibrosis. Front Biosci. 7: d793–d807.
Powell EE, Edwards-Smith CJ, Hay JL, Clouston AD, Crawford DH, Shorthouse C, Purdie DM & Jonsson JR (2000) Host genetic factors influence disease progression in chronic hepatitis C. Hepatology 31: 828–833.
Belli LS, Zavaglia C, Alberti AB, Poli F, Rondinara G, Silini E, Taioli E, de Carlis L, Scalamogna M, Forti D, Pinzello G & Ideo G (2000) Influence of immunogenetic background on the outcome of recurrent hepatitis C after liver transplantation. Hepatology 31: 1345–1350.
Britton RS & Bacon BR (2002) Hereditary hemochromatosis and alcohol: a fibrogenic cocktail. Gastroenterology 122: 563–565.
Fletcher LM, Dixon JL, Purdie DM, Powell LW & Crawford DH (2002) Excess alcohol greatly increases the prevalence of cirrhosis in hereditary hemochromatosis. Gastroenterology 122: 281–289.
Piperno A, Fargion S, D'Alba R, Roffi L, Fracanzani AL, Vecchi L, Failla M & Fiorelli G (1992) Liver damage in Italian patients with hereditary hemochromatosis is highly influenced by hepatitis B and C virus infection. J. Hepatol. 16: 364–368.
Monto A, Alonzo J, Watson JJ, Grunfeld C & Wright TL (2002) Steatosis in chronic hepatitis C: Relative contributions of obesity, diabetes mellitus, and alcohol. Hepatology 36: 729–736.
Tilg H & Diehl AM (2000) Cytokines in alcoholic and nonalcoholic steatohepatitis. N. Engl. J. Med. 343: 1467–1476.
Falck-Ytter Y, Younossi ZM, Marchesini G & McCullough AJ (2001) Clinical features and natural history of nonalcoholic steatosis syndromes. Semin. Liver Dis. 21: 17–26.
Wiese M, Berr F, Lafrenz M, Porst H & Oesen U (2000) Low frequency of cirrhosis in a hepatitis C (genotype 1b) single-source outbreak in Germany: a 20-year multicenter study. Hepatology 32: 91–96.
Barrett S, Goh J, Coughlan B, Ryan E, Stewart S, Cockram A, O'Keane JC & Crowe J (2001) The natural course of hepatitis C virus infection after 22 years in a unique homogenous cohort: spontaneous viral clearance and chronic HCV infection. Gut 49: 423–430.
Alter HJ & Seeff LB (2000) Recovery, Persistence, and Sequelae in Hepatitis C Virus Infection: A Perspective on Long-Term Outcome. Sem. Liver Dis. 20: 17–35.
Mallat A, Preaux AM, Blazejewski S, Rosenbaum J, Dhumeaux D & Mavier P (1995) Interferon alfa and gamma inhibit proliferation and collagen synthesis of human Ito cells in culture. Hepatology 21: 1003–1010.
Shiffman M, Hoffman CM, Contos MJ, Luketc VA, Sanyal AJ, Sterling RK, Ferreira-Gonzales A, Mills AS & Garret C (1999) A randomised controlled trial of maintenance interferon therapy for patients with chronic hepatitis C virus and persistent viremia. Gastroenterology 117: 1164–1172.
Schuppan D & Hahn EG (2000) Interleukin-10: The magic bullet for liver fibrosis?. Gastroenterology 119: 1412–1414.
Di Martino V, Rufat P, Boyer N, Renard P, Degot F, Martignot-Peignoux M, Matheron S, Le Moing V, Vachon F, Degott C, Valla D & Marcellin P (2001) The influence of human immunodeficiency virus coinfection on chronic hepatitis C in injection drug users: a long-term retrospective cohort study. Hepatology 34: 1193–1199.
Kamal SM, Rasenack JB, Bianci L, Al Tawil A, El Sayed Khalifa K, Peter T, Mansour H, Ezzat W & Koziel M (2001) Acute hepatitis C without and with Shistosomiasis: correlation with hepatitis C-specific CD4+ T-cell and cytokine response. Gastroenterology 121: 646–656.
Pietschmann T, Lohmann V, Kaul A, Krieger N, Rinck G, Rutter G, Strand D & Bartenschlager R (2002) Persistent and transient replication of full-length hepatitis C virus genomes in cell culture. J. Virol. 76: 4008–4021.
Mercer DF, Schiller DE, Elliott JF, Douglas DN, Hao C, Rinfret A, Addison WR, Fischer KP, Churchill TA, Lakey JR, Tyrrell DL & Kneteman NM (2001) Hepatitis C virus replication in mice with chimeric human livers. Nat. Med. 7: 927–933.
Lai MM (2002) Hepatitis C virus proteins: direct link to hepatic oxidative stress, steatosis, carcinogenesis and more. Gastroenterology 122: 568–571.
Lo SY, Selby MJ & Ou JH (1996) Interaction between hepatitis C virus core protein and E1 envelope protein. J. Virol. 70: 5177–5182.
Moriya K, Yotsuyanagi H, Shintani Y, Fujie H, Ishibashi K, Matsuura Y, Miyamura T & Koike K (1997) Hepatitis C virus core protein induces hepatic steatosis in transgenic mice. J. Gen. Virol. 78: 1527–1531.
Moriya K, Fujie H, Shintani Y, Yotsuyanagi H, Tsutsumi T, Ishibashi K, Matsuura Y, Kimura S, Miyamura T & Koike K (1998) The core protein of hepatitis C virus induces hepatocellular carcinoma in transgenic mice. Nat. Med. 4: 1065–1067.
Basu A, Meyer K, Ray RB & Ray R (2002) Hepatitis C virus core protein is necessary for the maintenance of immortalized human hepatocytes. Virology 298: 53–62.
Zhu N, Khoshnan A, Schneider R, Matsumoto M, Dennert G, Ware C & Lai MM (1998) Hepatitis C virus core protein binds to the cytoplasmic domain of tumor necrosis factor (TNF) receptor 1 and enhances TNF-induced apoptosis. J. Virol. 72: 3691–3697.
You LR, Chen CM & Lee YH (1999) Hepatitis C virus core protein enhances NF-kappaB signal pathway triggering by lymphotoxin-beta receptor ligand and tumor necrosis factor alpha. J. Virol 73: 1672–1681.
Otsuka M, Kato N, Lan K, Yoshida H, Kato J, Goto T, Shiratori Y & Omata M (2000) Hepatitis C virus core protein enhances p53 function through augmentation of DNA binding affinity and transcriptional ability. J. Biol. Chem. 275: 34122–34130.
Ray RB, Steele R, Meyer K & Ray R (1998) Hepatitis C virus core protein represses p21WAF1/Cip1/Sid1 promoter activity. Gene 208: 331–336.
Yang SH, Lee CG, Lee CW, Choi EJ, Yoon SK, Ahn KS & Sung YC (2002) Hepatitis C virus core inhibits the Fas-mediated p38 mitogen activated kinase signaling pathway in hepatocytes. Mol. Cells 13: 452–462.
Otsuka M, Kato N, Taniguchi H, Yoshida H, Goto T, Shiratori Y & Omata M (2002) Hepatitis C virus core protein inhibits apoptosis via enhanced Bcl-xL expression. Virology 296: 84–93.
Perlemuter G, Sabile A, Letteron P, Vona G, Topilco A, Chretien Y, Koike K, Pessayre D, Chapman J, Barba G & Brechot C (2002) Hepatitis C virus core protein inhibits microsomal triglyceride transfer protein activity and very low density lipoprotein secretion: a model of viral-related steatosis. FASEB. J. 16: 185–194.
Tsutsumi T, Suzuki T, Shimoike T, Suzuki R, Moriya K, Shintani Y, Fujie H, Matsuura Y, Koike K & Miyamura T (2002) Interaction of hepatitis C virus core protein with retinoid X receptor alpha modulates its transcriptional activity. Hepatology 35: 937–946.
Okuda M, Li K, Beard MR, Showalter LA, Scholle F, Lemon SM & Weinman SA (2002) Mitochondrial injury, oxidative stress, and antioxidant gene expression are induced by hepatitis C virus core protein. Gastroenterology 122: 366–375.
Moriya K, Nakagawa K, Santa T, Shintani Y, Fujie H, Miyoshi H, Tsutsumi T, Miyazawa T, Ishibashi K, Horie T, Imai K, Todoroki T, Kimura S & Koike K (2001) Oxidative stress in the absence of inflammation in a mouse model for hepatitis C virus-associated hepatocarcinogenesis. Cancer Res 61: 4365–4370.
Gale, Jr M, Kwieciszewski B, Dossett M, Nakao H & Katze MG (1999) Antiapoptotic and oncogenic potentials of hepatitis C virus are linked to interferon resistance by viral repression of the PKR protein kinase. J Virol 73: 6506–6516.
Francois C, Duverlie G, Rebouillat D, Khorsi H, Castelain S, Blum HE, Gatignol A, Wychowski C, Moradpour D & Meurs EF (2000) Expression of hepatitis C virus proteins interferes with the antiviral action of interferon independently of PKR-mediated control of protein synthesis. J. Virol. 74: 5587–5596.
Gong G, Waris G, Tanveer R & Siddiqui A (2001) Human hepatitis C virus NS5A protein alters intracellular calcium levels, induces oxidative stress, and activates STAT-3 and NF-kappa B. Proc Natl Acad Sci USA 98: 9599–9604.
Arima N, Kao CY, Licht T, Padmanabhan R, Sasaguri Y & Padmanabhan R (2001) Modulation of cell growth by the hepatitis C virus nonstructural protein NS5A. J. Biol. Chem. 276: 12675–12684.
Lerat H, Honda M, Beard MR, Loesch K, Sun J, Yang Y, Okuda M, Gosert R, Xiao SY, Weinman SA & Lemon SM (2002) Steatosis and liver cancer in transgenic mice expressing the structural and nonstructural proteins of hepatitis C virus. Gastroenterology 122: 352–365.
Bismut FI, Ratziu V, Pieroni L, Charlotte F, Benhamou Y & Poynard T (2001) Biochemical markers of liver fibrosis in patients with chronic hepatitis C virus infection: a prospective study:. Lancet 357: 1069–1075.
Schuppan D, Stölzel U, Oesterling C & Somasundaram R (1995) Serum markers for liver fibrosis. J Hepatol. 22: Suppl 2: 82–88.
Poniachik J, Bernstein DE, Reddy KR, Jeffers LJ, Coelho-Little ME, Civantos F & Schiff ER (1996) The role of laparoscopy in the diagnosis of cirrhosis. Gastrointest. Endosc. 43: 568–571.
Qi Z, Atsuchi N, Ooshima A, Takeshita A & Ueno H (1999) Blockade of type beta transforming growth factor signaling prevents liver fibrosis and dysfunction in the rat. Proc Natl Acad Sci USA 96: 2345–2349.
George J, Roulot D, Koteliansky VE & Bissell DM (1999) In vivo inhibition of rat stellate cell activation by soluble transforming growth factor beta type II receptor: a potential new therapy for hepatic fibrosis. Proc Natl Acad Sci USA 96: 12719–12724.
Yata Y, Gotwals P, Koteliansky V & Rockey DC (2002) Dose-dependent inhibition of hepatic fibrosis in mice by a TGF-beta soluble receptor: implications for antifibrotic therapy. Hepatology 35: 1022–1030.
Mori T, Kawara S, Shinozaki M, Hayashi N, Kakinuma T, Igarashi A, Takigawa M, Nakanishi T & Takehara K (1999) Role and interaction of connective tissue growth factor with transforming growth factor-beta in persistent fibrosis: A mouse fibrosis model. J. Cell. Physiol. 181: 153–159.
Ueki T, Kaneda Y, Tsutsui H, Nakanishi K, Sawa Y, Morishita R, Matsumoto K, Nakamura T, Takahashi H, Okamoto E & Fujimoto J (1999) Hepatocyte growth factor gene therapy of liver cirrhosis in rats. Nat. Med. 5: 226–230.
Jia JD, Bauer M, Ruehl M, Milani S, Boigk G, Riecken EO & Schuppan D (2001) Antifibrotic effect of silymarin in rat secondary biliary fibrosis is mediated by downregulation of procollagen I, TIMP-1 and TGF-β1 RNA. J. Hepatol. 35: 392–398.
Shimizu I, Ma YR, Mizobuchi Y, Liu F, Miura T, Nakai Y, Yasuda M, Shiba M, Horie T, Amagaya S, Kawada N, Hori H & Ito S (1999) Effects of Sho-saiko-to, a Japanese herbal medicine, on hepatic fibrosis in rats. Hepatology 29: 282–284.
Bruck R, Genina O, Aeed H, Alexiev R, Nagler A, Avni Y & Pines M (2001) Halofuginone to prevent and treat thioacetamide-induced liver fibrosis in rats. Hepatology 33: 379–386.
De Bleser PJ, Xu G, Rombouts K, Rogiers V & Geerts A (1999) Glutathione levels discriminate between oxidative stress and transforming growth factor-beta signaling in activated rat hepatic stellate cells. J. Biol. Chem. 274: 33881–33887.
Garcia-Trevijano ER, Iraburu MJ, Fontana L, Dominguez-Rosales JA, Auster A, Covarrubias-Pinedo A & Rojkind M (1999) Transforming growth factor beta1 induces the expression of alpha1(I) procollagen mRNA by a hydrogen peroxide-C/EBPbeta-dependent mechanism in rat hepatic stellate cells. Hepatology 29: 960–970.
Duncan MR, Hasan A & Berman B (1995) Pentoxifylline, pentifylline, and interferons decrease type I and III procollagen mRNA levels in dermal fibroblasts: evidence for mediation by nuclear factor 1 down-regulation. J. Invest. Dermatol. 104: 282–286.
Raetsch C, Boigk G, Herbst H, Riecken EO & Schuppan D (2002) Pentoxifylline retards collagen accumulation in early but not in advanced rat secondary biliary fibrosis. Gut 50: 241–247.
Cho JJ, Hocher B, Herbst H, Jia JJ, Boigk G, Hahn EG, Riecken EO & Schuppan D (2000) An oral endothelin A receptor antagonist blocks collagen synthesis and deposition in advanced rat secondary fibrosis. Gastroenterology 118: 1169–1178.
Jonsson JR, Clouston AD, Ando Y, Kelemen LI, Horn MJ, Adamson MD, Purdie DM & Powell EE (2001) Angiotensin-converting enzyme inhibition attenuates the progression of rat hepatic fibrosis. Gastroenterology 121: 148–155.
Paizis G, Gilbert RE, Cooper ME, Murthi P, Schembri JM, Wu LL, Rumble JR, Kelly DJ, Tikellis C, Cox A, Smallwood RA & Angus PW (2001) Effect of angiotensin II type 1 receptor blockade on experimental hepatic fibrogenesis. J. Hepatol. 35: 376–385.
Somasundaram R, Ruehl M, Tiling N, Schmid M, Ackermann R, Riecken EO & Schuppan D (2000) Collagens serve as an extracellular store of bioactive interleukin 2. J. Biol. Chem. 275: 38170–38175.
Somasundaram R, Ruehl M, Schaefer B, Schmid M, Ackermann R, Riecken EO, Zeitz M & Schuppan D (2002) Interstitial collagens I, III and VI sequester and modulate the multifunctional cytokine oncostatin M. J. Biol. Chem. 277: 3242–3246.
Ruehl M, Somasundaram R, Schoenfelder I, Farndale RW, Knight CG, Schmid M, Ackermann R, Riecken EO, Zeitz M & Schuppan D (2002) The epithelial mitogen keratinocyte growth factor binds to collagens via the consensus sequence glycine-proline-hydroxyproline. J. Biol. Chem. 277: 26872–26878.
Grinnell F (1994) Fibroblasts, myofibroblasts and wound contraction. J. Cell. Biol. 124: 401–404.
Lin YC, Ho CH & Grinnell F (1998) Decreased PDGF receptor kinase activity in fibroblasts contracting stressed collagen matrices. Exp. Cell Res. 240: 377–387.
Pinzani M, Milani S, Herbst H, DeFranco R, Grappone C, Gentilini A, Caligiuri A, Pellegrini G, Ngo DV, Romanelli RG & Gentilini P (1996) Expression of platelet-derived growth factor and its receptors in normal human liver and during active hepatic fibrogenesis. Am. J. Pathol. 148: 785–800.
Ruehl M, Johannsen M, Atkinson J, Manski D, Sahin E & Schuppan D (1999) Soluble collagen VI induces tyrosine-phosphorylation of paxillin and focal adhesion kinase and activates the MAP-kinase erk2 in fibroblasts. Exp. Cell Res. 250: 548–557.
Beljaars L, Molema G, Schuppan D, Geerts A, deBleser P, Weert B, Meijer DKF & Poelstra K (2000) Successful targeting to rat hepatic stellate cells using albumin modified with cyclic peptides that recognize the collagen type VI receptor. J. Biol. Chem. 275: 12743–12751.
Author information
Authors and Affiliations
Corresponding author
Additional information
Edited by M Piacentini
Rights and permissions
About this article
Cite this article
Schuppan, D., Krebs, A., Bauer, M. et al. Hepatitis C and liver fibrosis. Cell Death Differ 10 (Suppl 1), S59–S67 (2003). https://doi.org/10.1038/sj.cdd.4401163
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.cdd.4401163
Keywords
This article is cited by
-
Association of TGF-ß1 polymorphisms and chronic hepatitis C infection: a Meta-analysis
BMC Infectious Diseases (2019)
-
Fibrogenic Gene Expression in Hepatic Stellate Cells Induced by HCV and HIV Replication in a Three Cell Co-Culture Model System
Scientific Reports (2019)
-
Tolloid-like 1 genetic variants determine fibrosis regression in chronic hepatitis C patients with curative antivirals
Scientific Reports (2018)
-
Signalome-wide assessment of host cell response to hepatitis C virus
Nature Communications (2017)
-
Activation of hepatic stellate cells by the ubiquitin C-terminal hydrolase 1 protein secreted from hepatitis C virus-infected hepatocytes
Scientific Reports (2017)
