
Abstract
Apoptosis of inflammatory cells is a critical event in the resolution of inflammation, as failure to undergo this form of cell death leads to increased tissue damage and exacerbation of the inflammatory response. Many factors are able to influence the rate of apoptosis in neutrophils, eosinophils, monocytes and macrophages. Among these is the signalling molecule nitric oxide (NO), which possesses both anti- and proapoptotic properties, depending on the concentration and flux of NO, and also the source from which NO is derived. This review summarises the differential effects of NO on inflammatory cell apoptosis and outlines potential mechanisms that have been proposed to explain such actions.
Similar content being viewed by others

Introduction
The free radical nitric oxide (NO) was first discovered as an endogenous vasodilator released from the endothelium to regulate vascular tone.1 However, it is now known that NO is a key mediator in a great number of physiological and pathophysiological processes (see Quinn).2 This ubiquitous signalling molecule can regulate the rate of apoptosis, or programmed cell death, in many cell types, including human inflammatory cells. Whether or not cells undergo apoptosis depends on the net balance of a large number of pro- versus antiapoptotic factors. Studies have revealed that NO has both pro and antiapoptotic properties, depending largely on the concentration and flux of NO, and the cell type under scrutiny (for reviews, see Nicotera et al.3 and Kim et al.4). It has been proposed that low concentrations of NO, derived from constitutively active endothelial and neuronal isoforms of NO synthase (eNOS and nNOS), usually have a protective effect on cells whereas higher concentrations derived from the inducible isoform (iNOS) are more likely to drive cell death.3
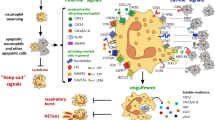
In stark contrast to cells undergoing necrosis, apoptotic inflammatory cells fail to release their proinflammatory and histotoxic contents.5 Furthermore, their clearance by professional phagocytes such as macrophages occurs via a nonphlogistic mechanism, which additionally aids the resolution of the inflammatory response (see Figure 1).6 Thus, apoptosis is generally regarded to be noninflammatory and is crucial for the successful resolution of inflammation. Chronic inflammatory conditions are frequently characterised by an apparent failure of myelocytic inflammatory cells to respond to apoptotic stimuli, or of phagocytes to remove apoptotic cells. Persistence of these cells leads to damage of the surrounding tissue and exacerbation of inflammation, as cells ultimately undergo either primary or secondary (following failed clearance of apoptotic cells) necrosis, an extremely proinflammatory form of cell death (see Figure 1).5,7 Manipulation of the rate of apoptosis in critical inflammatory effector cells, such as neutrophils, eosinophils, monocytes and macrophages, could therefore be of therapeutic benefit.7 NO is capable of inducing inflammatory cell apoptosis and also possesses several other anti-inflammatory properties, including direct downregulation of leukocyte functions, such as neutrophil and monocyte adhesion, and neutrophil chemotaxis, degranulation and superoxide anion (O2−) generation.8,9,10 It also acts to maintain the impermeable nature of the vascular endothelium to leukocytes.9 Thus, manipulation of NO concentrations is a particularly promising candidate to alter leukocyte function and rates of apoptosis in inflammatory conditions.

Death pathways and phagocytic clearance of inflammatory cells. Cells may undergo apoptosis, the noninflammatory form of cell death, in which granule contents are retained within the cell, and cells are cleared by macrophages that release anti-inflammatory mediators. Alternatively, they may undergo primary necrosis, during which the cell membrane integrity is compromised and granule contents are lost, exacerbating inflammation and tissue injury. Phagocytosis of these cells leads to release of proinflammatory mediators from the macrophages. In the event of failure of phagocytosis, or if the capacity of the phagocytes to ingest cells is exceeded, then apoptotic cells may enter into secondary necrosis. Again, this leads to release of toxic granule contents and clearance by a proinflammatory mechanism. TGF, transforming growth factor; IL, interleukin; TNF, tumour necrosis factor; Tx, thromboxane
A number of different NO donor classes have been utilised to investigate the role of NO and NO-related species in apoptotic processes. These fall into several broad categories defined by the species derived and the metabolic process underlying NO release.11 Existing drugs such as sodium nitroprusside (SNP), azide and hydroxylamine require complex metabolism to generate intracellular NO. Release of NO from azide and hydroxylamine is catalase-dependent, while membrane-bound proteins are thought to have a role in NO generation from SNP.11,12 Another class of NO donors is the S-nitrosothiols, which can release NO spontaneously, probably both inside and outside the cell. However, they also have the ability to transfer NO+ to free reduced thiols and cysteine residues in proteins, thus modulating enzyme activity, and this may constitute an important component of their cellular effects.12,13 The diazeniumdiolate, or ‘NONOate’, compounds are nucleophiles with two molecules of NO, which are spontaneously released in aqueous solution in a temperature- and pH-dependent reaction. A wide variety of diazeniumdiolates are available, such as spermine (SPER/NO), diethylamine (DEA/NO) and diethylenetriamine (DETA/NO)-based compounds, each with a different rate of release of NO that depends on the nature of the nucleophile.14 Finally, although not strictly NO donors, the sydnonimines are frequently used in studies into the effects of NO. These compounds, such as SIN-1, generate equal amounts of NO and O2−, which combine rapidly to form peroxynitrite (ONOO−); as a result, they are generally considered to be ONOO− donors.15 As the oxatriazole-5-imine derivative GEA 3162 is structurally similar to molsidomine, the precursor of SIN-1, it is possible that GEA 3162 generates ONOO− rather than pure NO, although this issue remains to be resolved.
This review will examine our current understanding of the regulation of inflammatory cell apoptosis by NO and the mechanisms through which the process is mediated. Primary human inflammatory cells will constitute the main focus of the review since cell lines are usually transformed and, therefore, their apoptotic machinery is likely to be altered. The biology and chemistry of NO,2 the various classes of NO donor11 and the apoptotic process itself16 have all previously been extensively reviewed elsewhere and will not be covered in detail here. The regulation of apoptosis in distinct inflammatory cells of the myeloid lineage will be considered first, and summarised in Table 1, then potential mechanisms through which NO may be acting will be discussed in general (Figure 2), as inevitably there seems to be a large degree of overlap between the mechanisms engaged in different cell types.

Proposed mechanisms for the effects of NO, its metabolites or by-products on inflammatory cell apoptosis. Antiapoptotic effects are thought to be mediated through an increase in cGMP, S-nitrosation of apoptotic effector proteins or activation of the transcription factor NF-κB by NO or reactive higher oxides of nitrogen, especially N2O3 the principal metabolite of NO which is likely to mediate many of its biological effects. A number of mechanisms have been suggested to explain the proapoptotic effects, which appear to converge on the release of cytochrome c from mitochondria and subsequent activation of caspases, the central proteases of the apoptotic cascade. NF-κB, nuclear factor kappa B; cGMP, cyclic GMP; ER, endoplasmic reticulum; ΔΨm, change in mitochondrial membrane potential; MPTP, mitochondrial permeability transition pore
Regulation of Inflammatory Cell Apoptosis by NO
Myelomonocytic cells
It is well established that exogenous or endogenous iNOS-derived NO induces apoptosis in macrophages. Indeed, the macrophage was the first cell type in which NO-mediated apoptosis was demonstrated.17,18 Much of the work on this cell type has been carried out using cell lines, in particular, the murine macrophage cell line RAW 264.7. However, uncertainties have now been cast over the relevance of data obtained with animal cell lines to humans, as it has become apparent that species differences are more important than initially anticipated. Critical differences have been discovered in enzyme pathways in macrophages from different species, which may have a profound effect on NO production in these cells. It has been demonstrated that, whereas murine- and bovine-stimulated macrophages can produce copious amounts of NO, human, caprine, lapine and porcine cells may not be able to do so.19,20 Thus, although it is clear that stimulation of murine macrophages with LPS and IFN-γ provokes NO production from iNOS, which is sufficient to induce apoptosis,21,22,23,24 it is by no means certain that this is also the case in human cells. Nevertheless, despite the relative inability of human macrophages to produce endogenous NO upon external stimulation, they appear to respond to exogenous sources of NO in much the same way as those from other species. NO from the S-nitrosothiol, S-nitrosoglutathione (GSNO) and the spontaneous NO generator, SPER/NO, increased apoptosis in both RAW 264.7 cells and primary human monocyte-derived macrophages.25 Therefore, observations using NO donors in murine cells could still be extrapolated to human cells, and may provide useful guidance on the potential therapeutic benefits of using NO in inflammatory conditions. Several NO donors have been demonstrated to elicit apoptosis in RAW 264.7 cells, including sodium nitroprusside (SNP),26 S-nitroso-N-acetylpenicillamine (SNAP),24 GSNO21,22,23,26,27,28 and the NONOates SPER/NO, 21,22,26,27,29 DEA/NO and DETA/NO.26
In addition, Sandoval et al.30 demonstrated increased apoptosis in RAW 264.7 cells upon exposure to peroxynitrite (ONOO−; 100–300 μM over 4 h and 10–100 μM over 14 h). Conversely, Scivittaro et al.27 observed that lower concentrations (30–50 μM) of ONOO− had a protective effect against LPS and IFN-γ-induced apoptosis in these cells, although not against exogenous NO-mediated apoptosis. This discrepancy may well be the result of the different concentrations of ONOO− used in the above studies. In addition, Brune et al.22 reported that RAW 264.7 cells that have higher levels of O2− are resistant to iNOS-mediated apoptosis, suggesting that ONOO− plays no role in mediating NO-induced apoptosis in murine macrophages, and indeed may have a protective role. Although Brockhaus and Brune31 showed that overexpression of superoxide dismutase (SOD) in RAW 264.7 macrophages inhibited NO-mediated apoptosis, which implies a role for ONOO−, on further investigation they found this not to be the case. The ONOO− scavenger, uric acid, failed to block apoptosis elicited by the NO donors GSNO and SPER/NO, while abolishing that induced by SIN-1, thus ruling out ONOO− as a mediator of NO-induced apoptosis. Inhibition of murine monocyte/macrophage J774 cell apoptosis by NO has also been reported when O2− was scavenged using SOD.32 Boggs et al.29 found that a subset of RAW 264.7 cells that were resistant to NO-induced apoptosis had lower glutathione (GSH) levels and, therefore, higher oxidant status than nonresistant cells.
Little work has been carried out to examine the effects of NO on apoptosis in primary human monocytes that have not matured into macrophages. Adrie et al.33 demonstrated that the peroxynitrite donor, SIN-1 (0.25−2 mM), induced cell death (apoptosis and necrosis) in primary human monocytes from healthy volunteers, but that NO from S-nitroso-N-acetylpenicillamine (SNAP; 0.5−2 mM) was unable to do so. Muhl et al.34 used U937 cells and peripheral blood mononuclear cells from human volunteers, and demonstrated an induction of apoptosis using DETA/NO, which has also been shown by Ferret et al.35 to induce apoptosis in U937 and THP-1 cells. Some studies have also demonstrated NO-induced apoptosis in HL-60 cells, using DETA/NO, SNP and SNAP,36,37,38 while ONOO− from SIN-1 can also induce apoptosis in human and canine long-term marrow cultures and J774 cells.39 Borutaite et al.40 demonstrated increased caspase activation in J774 cells treated with GSNO, SNAP and DETA/NO (1mM), which is generally accepted to be an indication of apoptosis. However, morphological apoptosis was not measured in these studies.
Neutrophils
In recent years, it has become clear that NO also has the ability to induce apoptosis in neutrophils. Wong et al.41 demonstrated that the oxatriazole-5-imine derivative, GEA 3162, and SIN-1 increased the rate of apoptosis in human neutrophils. Simultaneously, Brennan et al.42 found increased markers of DNA fragmentation treated with high (8–32 mM) concentrations of SNP in these cells. However, this effect may have been the result of respiratory inhibition by cyanide derived from the liberation of NO from such high concentrations of this compound. Since then, a number of groups have validated these findings through the use of several different sources of NO. NO gas (20 and 50 ppm) reduces cell viability and augments DNA fragmentation over 2 or 24 h of culture, an effect that is particularly pronounced in the presence of 80% O2.43 Whether this cell death comprises apoptosis or merely necrosis, however, remains unclear. An induction of neutrophil apoptosis has also been observed using both traditional NO donors such as SNP44 and different sources of NO such as GSNO (0.5–5 mM).45 Blaylock et al.46 reported that GEA 3162 (10 μM) increased neutrophil apoptosis as assessed by annexin V binding to exposed phosphatidylserine (PS) at 4 and 8 h, but not at 16 h. On the other hand, the ONOO− donor, SIN-1 (1 mM), showed no significant increase in PS exposure compared to control cells, although there was a small enhancement of annexin V binding at 4 h. The effects of these two compounds were also studied by Ward et al.,10 who demonstrated that concentrations of 10–100 μM GEA 3162 and 0.3–3 mM SIN-1 enhanced caspase-dependent morphological neutrophil apoptosis at 6 and 20 h, and 30–100 mM GEA 3162 increased annexin V binding at 6 h. In contrast to Blaylock's study, however, the increase in morphological apoptosis by 10 μM GEA 3162 was not significant at 6 h, whereas Blaylock observed significant differences at just 4 h. The principal methodological difference between the two studies is in the culture conditions. It has previously been demonstrated that the microenvironment, including cell density and concentration of plasma proteins, has a critical effect on the rate of neutrophil apoptosis in vitro.47 It must therefore be emphasised that the choice of culture conditions may subtly alter the effects of NO on neutrophils, and experimental design should receive careful consideration.
The early induction of PS exposure, but later absence of differences between control and GEA 3162-treated cells was confirmed and extended by Taylor et al.48 in 2001. It was reported that 30–100 μM GEA 3162, 300 μM–1 mM SPER/NO and 1 mM DEA/NO all induced morphological evidence of neutrophil apoptosis over 20 h. However, PS exposure was only increased from control levels at early time points in GEA 3162-treated neutrophils. The shedding of CD16 by apoptotic neutrophils equated well with morphological indications of apoptosis, but DNA fragmentation, as evidenced by propidium iodide intercalation and DNA ladders, was not necessarily observed alongside other markers of apoptosis. GEA 3162 (30–100 μM) and SPER/NO (1 mM), but not DEA/NO (1 mM), produced a paradoxical inhibition of DNA fragmentation despite a clear induction of apoptosis as assessed by other techniques.
Although there is a large body of evidence to demonstrate the proapoptotic effect of NO on neutrophils and the antiapoptotic effects in many cell types, it was not until recently that inhibition of neutrophil apoptosis by NO was reported.48 NO derived spontaneously from low concentrations of the diazeniumdiolate compounds SPER/NO and DEA/NO reduced the rate of neutrophil apoptosis over a 20-h time course. In contrast, equivalent concentrations of the oxatriazole derivative, GEA 3162, produced no such inhibition, suggesting that the antiapoptotic effects of NO may depend on the mechanism of NO liberation from donor drugs.
Eosinophils
Conflicting results have been reported for the effects of exogenous NO on eosinophil apoptosis. Beauvais et al.49 reported in 1995 that the catalase-dependent NO donors, azide and hydroxylamine, dramatically reduced the rate of apoptosis in this cell type, as evidenced by several apoptotic markers, with optimum concentrations for these effects being 100 μM for azide and 300 μM for hydroxylamine. In contrast, only a small inhibitory effect of azide (0.02–1.2 mM), hydroxylamine (300 μM–1.2 mM) and the S-nitrosothiol, SNAP (1 μM–1 mM), on constitutive eosinophil apoptosis was observed by Hebestreit et al.50 However, Beauvais et al.51 later published that other sources of NO, which are not dependent upon enzymatic activity to generate NO, fail to promote eosinophil survival. Although SNAP (1–5 mM) inhibits eosinophil apoptosis, it appears to drive the cells into a necrotic state; overall cell survival is virtually unchanged. Similar effects were also reported for SIN-1, but it appears that cell survival actually increased as expected on exposure to this compound, with a concomitant decrease in apoptosis. Treatment with S-nitroso-cysteine (SNOC), which has a very fast rate of NO release, produced little change in either cell survival or apoptosis. However, it is important to note that the concentrations used in this study were extremely high (1–5 mM) and it appears that lower concentrations of SNOC (1–3 mM) and SNAP (1 mM) may in fact slightly increase apoptosis. It is therefore possible that submillimolar concentrations could induce eosinophil apoptosis, but that higher concentrations are sufficiently toxic to push the cells into necrosis. The increased eosinophil survival observed with azide and hydroxylamine was mimicked when 3–10 μM SNAP was added to the cells in the presence of 10 μM haematin, the ferric form of haeme. On the other hand, when the cells were first primed using the cytokine IL-5 (which promotes eosinophil survival), a simultaneous inhibition of eosinophil survival and induction of apoptosis was observed with SNOC (100 μM–3 mM) and DETA/NO (300 μM–3 mM). This difference from unstimulated cells was put down to the priming effect of IL-5. However, the concentrations used in these studies were considerably lower than in nonprimed cells, and as previously mentioned, submillimolar concentrations of these compounds could conceivably be able to induce apoptosis rather than necrosis, whether or not the cells are primed.
Alongside spontaneous eosinophil apoptosis, NO has also been reported to inhibit cell death stimulated by Fas receptor activation. Hebestreit et al.50 demonstrated that enhanced DNA fragmentation and PS exposure provoked by treatment with anti-Fas monoclonal antibody could be attenuated by NO derived from LPS and IFN-γ-stimulated human monocyte-like U937 cells. This inhibitory effect on Fas-mediated apoptosis could be blocked using inhibitors of NOS (L-NMMA; 1 mM) or the GC pathway (LY 83583; 10 μM), and was mimicked in a concentration-dependent manner by exogenous NO from 0.02–1.2 mM azide, 0.3–1.2 mM hydroxylamine and 1 μM–1 mM SNAP.
In vivo effects of NO and its therapeutic potential
Delayed apoptosis of activated granulocytes has been reported to occur in a number of inflammatory conditions in humans or animal models,5,52 including rheumatoid arthritis,53 acute pancreatitis,54 bacterial pneumonia,55 inflammatory bowel disease,56 asthma,57,58,59 and following surgery.60,61 Failure of these cells to undergo programmed cell death and to be cleared by phagocytes allows persistent and inappropriate inflammation to occur, as activated granulocytes release a number of proinflammatory mediators, which may contribute significantly to the aetiology of the disease. Therefore, the induction of apoptosis in these cells is a potential target for therapeutic intervention, by removal of the inflammatory effector cells, thereby minimising tissue damage and oedema. As NO has a wide range of anti-inflammatory properties, including inhibition of platelet and neutrophil functions (see Granger and Kubes9 for review), this molecule could provide effective multitarget therapy against inflammation.
Although iNOS has been implicated in the pathogenesis of certain inflammatory diseases, such as arthritis, SLE and irritable bowel syndrome,62 a number of studies have demonstrated a protective effect of NO against several conditions characterised by inflammation, such as glomerulonephritis,63 acute hepatic necrosis,64 arthritis,65,66 endotoxaemia64 and acute lung injury67 in vivo (reviewed by Clancy and Abramson62). Most studies attribute these effects to the wide range of general anti-inflammatory properties of NO, as reviewed by Granger and Kubes,9 and are outwith the scope of this review. Of particular interest with regard to this review, however, is the increasing evidence that the protection afforded against inflammation and immunity by NO may be mediated in part through the induction of inflammatory cell apoptosis. As for apoptosis induction in vitro, the concentration of NO in the local environment,68,69 the timing of administration70,71 or the route of administration, and perhaps therefore the NOS isoform targeted72 appears to be critical. Studies have suggested that lower doses of NO may be detrimental but that higher doses may attenuate the inflammatory response, with some authors proposing a role for the cytotoxic effects of NO on myeloid inflammatory cells.69,73,74,75
NO appears to be particularly effective in autoimmune conditions, such as experimental allergic encephalomyelitis (EAE), which serves as a model for human multiple sclerosis.76 In this model, several studies have reported that iNOS-deficient rats or mice that were immunised directly with myelin basic protein developed exacerbated disease, although results obtained following immunisation with myelin basic protein-specific T cells often contradict these findings.76 One proposed mechanism for the protective effect of iNOS-derived NO is induction of apoptosis in macrophages or T cells.71,77
Other autoimmune disease models in which NO has been reported to be protective are the rat model of autoimmune interstitial nephritis and experimental autoimmune uveitis, as inhibition of NOS caused exacerbated injury in both models, although again other studies have produced conflicting results.76 Therefore, NO may be protective in a number of autoimmune conditions, although further research will be required to fully understand the apparent contradictory effects of NO.
A beneficial protective effect of NO has also been shown in the elicitation phase of contact hypersensitivity, as prolonged inflammatory reactions were observed in iNOS knockout mice.69 Again, it has been suggested that this effect may be partly because of the induction of apoptosis in infiltrating cells. In a human model similar to sunburn and psoriasis, Ormerod et al.68 described a cytotoxic effect of high concentrations of topically administered NO to immunocompetent cells, which was not seen with low concentrations. Aminoguanidine, an inhibitor of iNOS, prevented the impairment of renal vascular bed responses and reduced urine nitrate levels and apoptotic mononuclear cells in a rat model of experimental nephropathy.78 However, although inflammatory apoptosis might be expected to be beneficial, the effect of this form of cell death on prognosis was not studied within the late, sclerotic phase of the disease during this study. In the same model, Rangan et al.79 discovered that NOS inhibitors exacerbated progression of the disease, and tubulointerstitial injury was also found to be increased in the presence of NOS inhibitors in a model of thrombotic microangiopathy.80
Two in vivo studies were published in 1999, investigating the effects of NO on macrophage apoptosis in cardiovascular disease of cholesterol-fed rabbits. Niebauer et al.75 demonstrated that provision of L-arginine (the substrate for NOS) in drinking water could reduce the formation of inflammatory lesions following balloon angioplasty, while Wang et al.74 showed that it decreased existing atherosclerotic lesions, by inducing macrophage apoptosis.
Thus, there is a significant body of evidence to suggest that supplementation of NO may be beneficial in inflammatory diseases and that the induction of apoptosis in infiltrating cells may have a role in mediating this protection. However, these issues remain controversial, with much conflicting evidence. It is clear, however, that provision of NO may not be suitable for the treatment of all inflammatory conditions, and indeed may only be appropriate at certain stages of disease progression or disease provoked by a particular mechanism. Much further work is required to clarify these issues.
Mechanisms of action of NO
The mechanisms through which NO is able to both promote and delay inflammatory cell apoptosis still remain to be fully elucidated. However, it is generally believed that low concentrations tend to be antiapoptotic, acting through a rise in cyclic GMP or S-nitrosation of caspase enzymes. On the other hand, high concentrations of NO are generally more toxic, inducing either apoptosis or necrosis, with most reports suggesting that such actions occur independently of the sGC signalling pathway (see below).
Another possibility is that NO may indirectly interfere with alternative pathways that regulate inflammatory cell survival. For example, glucocorticoids (GCs) promote eosinophil apoptosis but inhibit neutrophil death.7,81 It has been shown that NO can inhibit GC receptor binding through S-nitrosation,82 and therefore could attenuate both the pro- and antiapoptotic effects of glucocorticoids on inflammatory cells.
Antiapoptotic mechanisms
The pathway by which inhibition of neutrophil apoptosis occurs has not yet been investigated. However, it is known that NO induces a rise in cGMP in neutrophils through activation of sGC.8,83 Given that the cell permeable analogues of cAMP (db-cAMP) and cGMP (db-cGMP) can delay constitutive neutrophil apoptosis,10 and that a rise in cGMP has been postulated to at least partially account for NO-mediated inhibition of apoptosis in other cell types,84,85 it is possible that an increase in one or other of these cyclic nucleotides mediates the inhibition of apoptosis in neutrophils exposed to low concentrations of NO. A role for cyclic nucleotide (cGMP or cAMP) signalling has also been proposed in NO-mediated inhibition of both constitutive and Fas-triggered eosinophil apoptosis. Beauvais et al.49 observed that NO could only reduce eosinophil apoptosis when compounds that form nitrosyl-haeme in their liberation of NO were used, which was mimicked by the permeable cGMP analogue db-cGMP and reduced by the sGC inhibitor LY 83583. Pure NO donors alone, in contrast, failed to increase eosinophil survival, although inhibition of apoptosis could be seen if haematin was added alongside a pure NO donor to artificially create a nitrosyl-haeme complex.51 It has been suggested that such inhibition occurs through activation of sGC by the nitrosyl-haeme complex,51 which has been shown to occur in vitro using purified sGC,86 but as yet there is no evidence to suggest that this may occur in intact cells in vivo. However, the mechanism is not fully understood: the issue is complicated by the fact that haeme groups are known to scavenge NO, and therefore might be expected to inhibit NO-mediated effects. Inhibition of Fas-mediated apoptosis was reproduced by both db-cGMP and db-cAMP, and potentiated by the phosphodiesterase inhibitor IBMX,50 again suggesting a role for cyclic nucleotides. This group localised the site of Fas receptor death pathway blockade to downstream of SMase activation and ceramide generation but upstream or around the level of JNK activation. So et al.87 showed inhibition of the stress protein JNK2 by NO in vitro via S-nitrosation.
Secondly, the transcription factor NF-κB is known to regulate neutrophil apoptosis as its inhibition leads to increased apoptosis.88 Agents such as LPS have been shown to delay apoptosis in this cell type through stimulation of NF-κB and subsequent caspase-1-dependent activation of IL-1β.89 It has been demonstrated in several cell types that activation of NF-κB leads to the transcriptional upregulation of survival factors such as the immediate early gene, IEX-1L, inhibitor of apoptosis proteins (IAPs), and members of the antiapoptotic Bcl-2 gene family, including Mcl-1, Bfl1/A1, Bcl-XL and Nr13, potentially explaining the survival effects of NO which, at low concentrations, induces NF-κB activation in macrophage cell lines90,91 and in human peripheral blood mononuclear cells.92 Activation of NF-κB may occur through a cGMP-dependent mechanism,93 and NO has been demonstrated to modulate the expression of several proteins affecting the activity of Bcl-2 family members through sGC activation. A cGMP-dependent mechanism has been proposed to account for the NO-induced downregulation of BNIP3, a dominant proapoptotic Bcl-2 family member in hepatocytes.94 Levels of MAP kinase phosphatase-3 (MKP-3) mRNA, which causes degradation of Bcl-2 via ERK1/2 dephosphorylation, were seen to be decreased by NO, thus protecting Bcl-2 levels and promoting survival of endothelial cells.95 The proapoptotic adapter protein, p66shc, is also downregulated by NO via sGC activation.96Furthermore, survival genes such as Bcl-2 and Bcl-XL have been shown to be upregulated in the presence of NO in endothelial cells97 and human neuroblastoma cells;96 therefore, it seems possible that there is a role for NF-κB-mediated transcriptional regulation in the antiapoptotic effects of NO in inflammatory cells.
Contrasting studies in macrophage cell lines22,29,32 suggest that the redox status of the cell may partially determine the effects of NO. An inhibitory effect of endogenous NO on J774 cell apoptosis can be unmasked when O2− is scavenged,32 and over-expression of SOD also protects RAW 264.7 cells against apoptosis induced by endogenous or exogenously supplied NO.31 These studies suggest a role for ONOO− in mediating NO-induced apoptosis. Conflicting evidence suggests that RAW 264.7 cells that overproduce O2− are resistant to NO-mediated apoptosis,22 and von Knethen et al.98 observed that O2− activates NF-κB, thus mediating survival in these cells. Furthermore, Brockhaus and Brune31 found that ONOO− had no role in NO-evoked apoptosis, despite the protective effect of SOD in RAW 264.7 cells. Others have found that such protection is observed when cellular thiols are depleted in RAW 264.7 cells.29 It has been reported that endogenous antioxidant levels,99 or the balance between oxidative and nitrosative stress,100 can determine the cellular response to NO. It has been proposed that in low thiol concentrations, NO actually protects against cell death, whereas it induces death in cells with normal thiol levels.99 Exogenous glutathione has also been shown to enhance neutrophil apoptosis and increase H2O2 levels, possibly leading to hydroxyl radical- mediated damage.101
In the absence of large quantities of scavenger thiols such as glutathione, but in the presence of oxygen, it is possible that NO S-nitrosates critical effector molecules of apoptosis such as caspases, thus preventing their activation and having an inhibitory effect on the proteolytic cascade. It has been shown by several groups that NO can inhibit a number of apoptotic proteins,102 including caspase 3 (the protease responsible for the initiation of internucleosomal DNA fragmentation),103,104,105,106,107,108 caspase 8,105,109,110 caspase 9,111 caspase 1109,110 and caspases 2, 3, 4, 6 and 7105 activation via S-nitrosation. Inhibition of caspase 3 has been reported to involve two distinct mechanisms in hepatocytes – direct protein S-nitrosation, and another mechanism, which has not yet been elucidated, but is dependent upon cGMP.108 Therefore, the cGMP-dependent antiapoptotic effects of NO in inflammatory cells may be mediated through an inhibitory effect on caspases.
Proapoptotic mechanisms
Studies have shown that apoptosis in neutrophils and macrophages proceeds via activation of caspase protease enzymes,10,112,113 part of the classical apoptotic effector cascade. However, the upstream mechanisms by which exposure to NO causes these enzymes to become activated has not been clarified, although several theories have been suggested.
ONOO− is considered to be one of the most likely candidates for the increased apoptosis of inflammatory cells observed with higher concentrations of NO, particularly in the case of neutrophils,10,43,46 which generate large quantities of O2− that rapidly combines with NO to form ONOO−. Compounds that generate ONOO−, such as SIN-1, may promote neutrophil apoptosis similar to that evoked by NO,10 and primary human monocytes also undergo apoptosis in response to ONOO− by a mechanism that involves mitochondrial membrane depolarisation, release of cytochrome c and caspase activation (see Figure 2).33 ONOO− has been shown to reversibly or irreversibly inhibit a number of mitochondrial respiratory complexes as well as inducing mitochondrial swelling, depolarisation, calcium release and permeability transition.114,115
Macrophage apoptosis can also be induced by exposure to ONOO−, via oxidative stress, which can be reduced by antioxidants such as ascorbic acid or phytolens.30,116 Induction of apoptosis by NO in elicited murine macrophages or RAW 264.7 cells has also been attributed to the formation of ONOO− within mitochondria, as nitrotyrosine residues were detected in cytochrome c.23 ONOO− and metabolites of NO (e.g. N2O3) can cause direct DNA damage or inhibit DNA repair enzymes,117 leading to an increase in the tumour suppressor protein p53, which has been shown to accumulate in NO-treated macrophages and may be the factor responsible for driving them towards apoptosis.22,25,118,119 The p53 protein promotes apoptosis through upregulation of the apoptotic proteins Bax and cyclin-dependent kinase p21, and downregulation of the antiapoptotic protein Bcl-2 (Figure 2).85 However, Gotoh et al.24 measured no increase of p53 in NO-mediated apoptosis in RAW 264.7 cells stimulated with LPS/IFN-γ. Instead, this group proposed a role for the endoplasmic reticulum stress pathway involving the transcription factors ATF6 and CHOP leading to cytochrome c release (Figure 2). Also, studies in murine macrophages31 suggest little or no involvement for ONOO− in NO-induced apoptosis in this cell type.
As previously described, the activation status of the survival factor, NF-κB, has been shown to play a role in regulation of the induction of inflammatory cell apoptosis.88 It has been demonstrated that high concentrations of NO can inhibit NF-κB activation in macrophage cell lines91 and human macrophages,120 monocytes121 and neutrophils.121,122 NO may inhibit NF-κB DNA binding through S-nitrosation of the p50 subunit of the transcription factor, as has been demonstrated in isolated NF-κB protein123 and in human respiratory cells and murine macrophages.124 Alternatively, transcriptional induction and stabilisation of the inhibitory molecule I-κB, that keeps NF-κB sequestered in the cytoplasm, may account for the inhibition of NF-κB activity.125 Furthermore, it has been reported that NO inhibits NF-κB activation in rat vascular smooth muscle cells via a cGMP-independent inhibition of the phosphorylation and proteasomal degradation of I- κB,126 and inhibition of the proteasome by NO has additionally been demonstrated by Glockzin et al.127 in macrophages. The result of such inhibition would be downregulation of survival factors under the control of this transcription factor, such as the antiapoptotic Bcl-2 family members. Indeed, this has been observed by a number of studies, as exogenous NO downregulates Bcl-2 but upregulates the proapoptotic protein, Bax, in neurons,128,129 and upregulates Bad and Bax, but downregulates Bcl-2 in human colon adenocarcinoma cells.130
In nonsmall cell lung cancer cells, it has been shown that NF-κB inhibition leads to apoptosis by increasing mitochondrial permeability, thus allowing release of cytochrome c and subsequent caspase activation (see Figure 2).131 This concurs with findings by Borutaite et al.,40 who demonstrated increased mitochondrial permeability and cytochrome c release from isolated rat mitochondria, and increased caspase activation in J774 cells treated with S-nitrosothiols (SNAP, GSNO) but not NONOates (DETA/NO), which activate caspases through an as yet unidentified alternative mechanism. As S-nitrosothiols readily transnitrosate endogenous cysteine residues, this supports the concept of S-nitrosation of the NF-κB p50 subunit as the mechanism of inhibition.
In addition, the biphasic effects of NO on NF-κB activation reported by Connelly et al. are mirrored by its effects on the open probability of the mitochondrial permeability transition pore (MPTP). Low concentrations of NO donors (GEA 3162, SNAP, SIN-1; 1–20μM) delayed or had no effect on MPTP opening, while at higher concentrations (20–100μM), opening was enhanced.132 In this study, GEA 3162 was found to be particularly effective at inducing MPTP opening compared to the other two NO donors, and this drug induces neutrophil apoptosis at lower concentrations than NONOates48 or SIN-1.10 Enhanced MPTP opening on exposure to NO was also reported by Hortelano et al.133 Therefore, there is growing evidence to suggest that the proapoptotic effects of NO in inflammatory cells may be mediated, at least in part, through inhibition of NF-κB.
Albina et al.18 proposed metabolic inhibition as a potential mechanism, as glucose starvation and inhibition of glycolysis or the TCA cycle all pushed macrophages into apoptosis. On the other hand, inhibition of the electron transport chain of respiration had no effect,18 and Messmer and Brune26 showed no reduction in NAD+ or ATP levels in NO-induced apoptosis, suggesting that NO does not act through respiratory inhibition. In contrast, others have reported that NO inhibits mitochondrial respiration through two distinct pathways.114,115 Reversible inhibition of cytochrome oxidase was seen with low concentrations of NO, whereas higher concentrations caused an inhibition of alternative respiratory chain complexes.115 Inhibition of complex IV was reversible, whereas inhibition of complex I was irreversible.114 Of course, the mechanisms described above may not be mutually exclusive. For example, respiratory inhibition by NO may enhance the production of reactive oxygen species by mitochondria, leading to the formation of ONOO− and providing an ONOO−-mediated pathway for NO-induced cytotoxicity (Figure 2).115
Conclusion
NO has a biphasic effect on apoptosis in many cell types, in which low concentrations delay but higher concentrations enhance this form of cell death, a pattern that has recently been confirmed in neutrophils. This correlates with the dichotomous action of NO on the activity of caspase enzymes responsible for the execution of apoptosis in vitro. Inhibition of caspases by S-nitrosation is a direct consequence of exposure to low concentrations of NO or, more likely, its oxidation products (e.g. N2O3). On the other hand, activation of these enzymes observed during the proapoptotic actions of higher concentrations represents a downstream event following initial effects on DNA or mitochondria, and can therefore be considered an indirect effect of NO.
Although the mechanism of inhibition has not yet been fully investigated, it is likely that cGMP production, NF-κB activation and subsequent expression of survival proteins or S-nitrosation of apoptotic proteins will play a major role. Inhibition of eosinophil apoptosis has been reported, but only with certain sources of NO that are capable of activating sGC with a consequent rise in cGMP. No such inhibitory effects have yet been demonstrated in monocytes or macrophages, and it remains to be seen whether these cell types are capable of producing such a response to low concentrations of NO.
It has been demonstrated that exogenous NO can induce apoptosis in all inflammatory cell types discussed in this review: monocytes, monocyte-derived macrophages, neutrophils and eosinophils. In addition, endogenous NO from iNOS also promotes apoptosis in macrophages. There still remains some controversy over the mechanism by which this molecule causes this form of cell death, although it involves activation of caspase proteases, and most agree that this occurs through a cGMP-independent pathway. Moreover, mitochondria appear to play a key role in the initiation of apoptosis by NO through release of cytochrome c, resulting in caspase activation.
It is clear that ONOO− derived from SIN-1 or other agents has the ability to promote apoptosis in its own right, but its role in mediating NO-induced apoptosis remains controversial. Some groups have reported that NO-evoked cytotoxicity is likely to be effected through ONOO− formation, while studies by others have indicated little or no role for ONOO−. Modulation of the activation status of the transcription factor NF-κB has also been proposed to account for NO-induced apoptosis in neutrophils and macrophages, and there is an increasing body of evidence to support this theory. On the other hand, DNA damage (by N2O3 or ONOO−) has also been shown, leading to an accumulation of the proapoptotic molecule p53.
Differences may exist in the mechanisms by which NO causes apoptosis in different cell types that could potentially be exploited to target a particular inflammatory cell type in certain conditions. Despite the uncertainties and controversies surrounding the regulation of inflammatory cell apoptosis by NO, it is clear that the class and concentration of NO-donating compound used and the cell type are critical determinants of the response. Major differences between different classes of NO donors and opposing effects with low and high concentrations of certain NO donors are observed. Thus, the amount and rate of NO release and the redox status of the target cell appear to be key factors in the cellular response to NO exposure, and certain NO donors appear to be more effective than others in promoting inflammatory cell apoptosis.
It is also important to realise that the concentration of NO donor used may not necessarily reflect the concentration of NO to which the cells are exposed. Equivalent concentrations of different NO donors may liberate NO to different extents or at different rates, or may produce different reactive nitrogen species, such as ONOO−. Culture conditions may also affect NO levels; for example, plasma proteins such as albumin are able to scavenge NO through the formation of S-nitrosothiols.13,134,135 Therefore, the concentration of free NO in the vicinity of the cells at any given time may vary from compound to compound, and the NO concentration in the system needs to be measured in order to directly compare different NO donors.
The vast majority of work on this subject has been carried out using in vitro systems, often utilising animal cell lines. How the results obtained in these systems relate to the in vivo situation during inflammation in humans still largely remains to be determined, but two studies in rabbits show that NO is a promising candidate for treatment or prevention of inflammatory conditions such as atherosclerosis and restenosis, possibly by influencing apoptosis.74,75 Further studies are required to elucidate completely the mechanism of action of NO on inflammatory cell apoptosis, in order to identify potential targets for the treatment of human inflammatory conditions and to evaluate the sources of NO that provide greatest therapeutic potential.
Abbreviations
- NO:
-
nitric oxide
- NOS:
-
nitric oxide synthase
- SNP:
-
sodium nitroprusside
- SPER/NO:
-
spermine diazeniumdiolate
- DEA/NO:
-
diethylamine diazeniumdiolate
- DETA/NO:
-
diethylenetriamine diazeniumdiolate
- SIN-1:
-
3-morpholino-sydnonimine
- O2−:
-
superoxide anion
- ONOO−:
-
peroxynitrite
- GEA 3162:
-
1,2,3,4-oxatriazolium, 5-amino-3-(3,4-dichorophenyl)-chloride
- LPS:
-
lipopolysaccharide
- IFN:
-
interferon
- GSNO:
-
S-nitrosoglutathione
- SNAP:
-
S-nitroso-N-acetylpenicillamine
- SOD:
-
superoxide dismutase
- GSH:
-
glutathione
- PS:
-
phosphatidylserine
- SNOC:
-
S-nitrosocysteine
- IL:
-
interleukin
- cGMP:
-
cyclic guanosine monophosphate
- sGC:
-
soluble guanylate cyclase
- GC:
-
glucocorticoid
- cAMP:
-
cyclic adenosine monophosphate
- NF-κB:
-
nuclear factor kappa B
- MPTP:
-
mitochondrial permeability transition pore
References
Palmer RM, Ferrige AG and Moncada S (1987) Nitric oxide release accounts for the biological activity of endothelium-derived relaxing factor. Nature 327: 524–526
Quinn AC, Petros AJ and Vallance P (1995) Nitric oxide: an endogenous gas. Br. J. Anaesth. 74: 443–451
Nicotera P, Brune B and Bagetta G (1997) Nitric oxide: inducer or suppressor of apoptosis?. Trends Pharmacol. Sci. 18: 189–190
Kim YM, Bombeck CA and Billiar TR (1999) Nitric oxide as a bifunctional regulator of apoptosis. Circ. Res. 84: 253–256
Haslett C (1997) Granulocyte apoptosis, and inflammatory disease. Br. Med. Bull. 53: 669–683
Meagher LC, Savill JS, Baker A, Fuller RW and Haslett C (1992) Phagocytosis of apoptotic neutrophils does not induce macrophage release of thromboxane B2. J. Leukoc. Biol. 52: 269–273
Ward C, Dransfield I, Chilvers ER, Haslett C and Rossi AG (1999) Pharmacological manipulation of granulocyte apoptosis: potential therapeutic targets. Trends Pharmacol. Sci. 20: 503–509
Moilanen E, Vuorinen P, Kankaanranta H, Metsa-Ketela T and Vapaatalo H (1993) Inhibition by nitric oxide donors of human polymorphonuclear leucocyte functions. Br. J. Pharmacol. 109: 852–858
Granger DN and Kubes P (1996) Nitric oxide as antiinflammatory agent. Methods Enzymol. 269: 434–442
Ward C, Wong TH, Murray J, Rahman I, Haslett C, Chilvers ER and Rossi AG (2000) Induction of human neutrophil apoptosis by nitric oxide donors: evidence for a caspase-dependent, cyclic-GMP-independent, mechanism. Biochem. Pharmacol. 59: 305–314
Megson IL (2000) Nitric oxide donor drugs. Drugs Fut. 25: 701–715
Feelisch M (1991) The biochemical pathways of nitric oxide formation from nitrovasodilators: appropriate choice of exogenous NO donors, and aspects of preparation, and handling of aqueous NO solutions. J. Cardiovasc. Pharmacol. 17(Suppl. 3): S25–S33
Butler AR and Rhodes P (1997) Chemistry, analysis, and biological roles of S-nitrosothiols. Anal. Biochem. 249: 1–9
Maragos CM, Morley D, Wink DA, Dunams TM, Saavedra JE, Hoffman A, Bove AA, Isaac L, Hrabie JA and Keefer LK (1991) Complexes of NO with nucleophiles as agents for the controlled biological release of nitric oxide. Vasorelaxant effects. J. Med. Chem 34: 3242–3247
Feelisch M, Ostrowski J and Noack E . (1989) On the mechanism of NO release from sydnonimines. J. Cardiovasc. Pharmacol. 14(Suppl 11): S13–S22
Haunstetter A and Izumo S (1998) Apoptosis: basic mechanisms, and implications for cardiovascular disease. Circ. Res. 82: 1111–1129
Sarih M, Souvannavong V and Adam A (1993) Nitric oxide synthase induces macrophage death by apoptosis. Biochem. Biophys. Res. Commun. 191: 503–508
Albina JE, Cui S, Mateo RB and Reichner JS (1993) Nitric oxide-mediated apoptosis in murine peritoneal macrophages. J. Immunol. 150: 5080–5085
Thomassen MJ and Kavuru MS (2001) Human alveolar macrophages, and monocytes as a source, and target for nitric oxide. Int. Immunopharmacol. 1: 1479–1490
Schneemann M and Schoedon G (2002) Species differences in macrophage NO production are important. Nat. Immunol. 3: 102
Messmer UK, Reed UK and Brune B (1996) Bcl-2 protects macrophages from nitric oxide-induced apoptosis. J. Biol. Chem. 271: 20192–20197
Brune B, Gotz C, Messmer UK, Sandau K, Hirvonen MR and Lapetina EG (1997) Superoxide formation, and macrophage resistance to nitric oxide-mediated apoptosis. J. Biol. Chem. 272: 7253–7258
Hortelano S, Alvarez AM and Bosca L (1999) Nitric oxide induces tyrosine nitration, and release of cytochrome c preceding an increase of mitochondrial transmembrane potential in macrophages. FASEB J. 13: 2311–2317
Gotoh T, Oyadomari S, Mori K and Mori M (2002) Nitric oxide-induced apoptosis in RAW 264.7 macrophages is mediated by endoplasmic reticulum stress pathway involving ATF6, and CHOP. J. Biol. Chem. 277: 12343–12350
von Knethen A, Brockhaus F, Kleiter I and Brune B (1999) NO-evoked macrophage apoptosis is attenuated by cAMP-induced gene expression. Mol. Med. 5: 672–684
Messmer UK and Brune B (1996) Nitric oxide (NO) in apoptotic Versus necrotic RAW 264.7 macrophage cell death: the role of NO-donor exposure, NAD+ content, and p53 accumulation. Arch. Biochem. Biophys. 327: 1–10
Scivittaro V, Boggs S, Mohr S and Lapetina EG (1997) Peroxynitrite protects RAW 264.7 macrophage from lipopolysaccharide/interferon-gamma-induced cell death. Biochem. Biophys. Res. Commun. 241: 37–42
Callsen D and Brune B (1999) Role of mitogen-activated protein kinases in S-nitrosoglutathione-induced macrophage apoptosis. Biochemistry 38: 2279–2286
Boggs SE, McCormick TS and Lapetina EG (1998) Glutathione levels determine apoptosis in macrophages. Biochem. Biophys. Res. Commun. 247: 229–233
Sandoval M, Zhang XJ, Liu X, Mannick EE, Clark DA and Miller MJ (1997) Peroxynitrite-induced apoptosis in T84, and RAW 264.7 cells: attenuation by L-ascorbic acid. Free Radic. Biol. Med. 22: 489–495
Brockhaus F and Brune B (1999) Overexpression of CuZn superoxide dismutase protects RAW 264.7 macrophages against nitric oxide cytotoxicity. Biochem. J. 338(Part 2): 295–303
Vouldoukis I, Sivan V, Vozenin MC, Kamate C, Calenda A, Mazier D and Dugas B (2000) Fc-receptor-mediated intracellular delivery of Cu/Zn-superoxide dismutase (SOD1) protects against redox-induced apoptosis through a nitric oxide dependent mechanism. Mol. Med. 6: 1042–1053
Adrie C, Richter C, Bachelet M, Banzet N, Francois D, Dinh-Xuan AT, Dhainaut JF, Polla BS and Richard MJ (2000) Contrasting effects of NO and peroxynitrites on HSP70 expression, and apoptosis in human monocytes. Am. J. Physiol. Cell Physiol. 279: C452–C460
Muhl H, Nold M, Chang JH, Frank S, Eberhardt W and Pfeilschifter J (1999) Expression and release of chemokines associated with apoptotic cell death in human promonocytic U937 cells, and peripheral blood mononuclear cells. Eur. J. Immunol. 29: 3225–3235
Ferret PJ, Soum E, Negre O, Wollman EE and Fradelizi D (2000) Protective effect of thioredoxin upon NO-mediated cell injury in THP1 monocytic human cells. Biochem. J. 346(Part 3): 759–765
Kuo ML, Chau YP, Wang JH and Shiah SG (1996) Inhibitors of poly (ADP-ribose) polymerase block nitric oxide-induced apoptosis but not differentiation in human leukemia HL-60 cells. Biochem. Biophys. Res. Commun. 219: 502–508
Kwak HJ, Jun CD, Pae HO, Yoo JC, Park YC, Choi BM, Na YG, Park RK, Chung HT, Chung HY, Park WY and Seo JS (1998) The role of inducible 70-kDa heat shock protein in cell cycle control, differentiation, and apoptotic cell death of the human myeloid leukemic HL-60 cells. Cell Immunol. 187: 1–12
Yabuki M, Tsutsui K, Horton AA, Yoshioka T and Utsumi K (2000) Caspase activation, and cytochrome c release during HL-60 cell apoptosis induced by a nitric oxide donor. Free Radic. Res. 32: 507–514
Lee JW, Beckham C, Michel BR, Rosen H and Deeg HJ (1997) HLA-DR-mediated signals for hematopoiesis, and induction of apoptosis involve but are not limited to a nitric oxide pathway. Blood 90: 217–225
Borutaite V, Morkuniene R and Brown GC (2000) Nitric oxide donors, nitrosothiols, and mitochondrial respiration inhibitors induce caspase activation by different mechanisms. FEBS Lett. 467: 155–159
Wong TH, Rossi AG and Chilvers ER (1997) Inhibition of neutrophil function, and survival by nitric oxide donors. Clin. Sci. 92: 7II
Brennan C, Stapleton PP, Redmond HP and Bouchier-Hayes D (1996) Nitric oxide-mediated neutrophil apoptosis. J. Leukoc. Biol. (Suppl): pp 39, A157.
Fortenberry JD, Owens ML, Brown MR, Atkinson D and Brown LA (1998) Exogenous nitric oxide enhances neutrophil cell death, and DNA fragmentation. Am. J. Respir. Cell Mol. Biol. 18: 421–428
Singhal PC, Patel P, Nahar N, Franki N, Kapasi A, Reddy K, Shah N, Nwakoby IE and Mehrotra B (1999) Ethanol-induced neutrophil apoptosis is mediated through nitric oxide. J. Leukoc. Biol. 66: 930–936
Fortenberry JD, Owens ML and Brown LA (1999) S-nitrosoglutathione enhances neutrophil DNA fragmentation, and cell death. Am. J. Physiol. 276: L435–L442
Blaylock MG, Cuthbertson BH, Galley HF, Ferguson NR and Webster NR (1998) The effect of nitric oxide, and peroxynitrite on apoptosis in human polymorphonuclear leukocytes. Free Radic. Biol. Med. 25: 748–752
Hannah S, Nadra I, Dransfield I, Pryde JG, Rossi AG and Haslett C (1998) Constitutive neutrophil apoptosis in culture is modulated by cell density independently of beta2 integrin-mediated adhesion. FEBS Lett. 421: 141–146
Taylor EL, Megson IL, Haslett C and Rossi AG (2001) Dissociation of DNA fragmentation from other hallmarks of apoptosis in nitric oxide-treated neutrophils: differences between individual nitric oxide donor drugs. Biochem. Biophys. Res. Commun. 289: 1229–1236
Beauvais F, Michel L and Dubertret L (1995) The nitric oxide donors, azide, and hydroxylamine, inhibit the programmed cell death of cytokine-deprived human eosinophils. FEBS Lett. 361: 229–232
Hebestreit H, Dibbert B, Balatti I, Braun D, Schapowal A, Blaser K and Simon HU (1998) Disruption of fas receptor signaling by nitric oxide in eosinophils. J. Exp. Med. 187: 415–425
Beauvais F and Joly F (1999) Effects of nitric oxide on the eosinophil survival in vitro. A role for nitrosyl-heme. FEBS Lett. 443: 37–40
Simon HU (1999) Apoptosis in inflammatory diseases. Int. Arch. Allergy Immunol. 118: 261–262
Ottonello L, Frumento G, Arduino N, Bertolotto M, Mancini M, Sottofattori E, Dallegri F and Cutolo M (2002) Delayed neutrophil apoptosis induced by synovial fluid in rheumatoid arthritis: role of cytokines, estrogens, and adenosine. Ann. N. Y. Acad. Sci. 966: 226–231
O'Neill S, O'Neill AJ, Conroy E, Brady HR, Fitzpatrick JM and Watson RW (2000) Altered caspase expression results in delayed neutrophil apoptosis in acute pancreatitis. J. Leukoc. Biol. 68: 15–20
Droemann D, Aries SP, Hansen F, Moellers M, Braun J, Katus HA and Dachoff K (2000) Decreased apoptosis, and increased activation of alveolar neutrophils in bacterial pneumonia. Chest 117: 1679–1684
Brannigan AE, O'Connell PR, Hurley H, O'Neill A, Brady HR, Fitzpatrick JM and Watson RW (2000) Neutrophil apoptosis is delayed in patients with inflammatory bowel disease. Shock 13: 361–366
Turlej RK, Fievez L, Sandersen CF, Dogne S, Kirschvink N, Lekeux P and Bureau F (2001) Enhanced survival of lung granulocytes in an animal model of asthma: evidence for a role of GM-CSF activated STAT5 signalling pathway. Thorax 56: 696–702
Woolley KL, Gibson PG, Carty K, Wilson AJ, Twaddell SH and Woolley MJ (1996) Eosinophil apoptosis, and the resolution of airway inflammation in asthma. Am. J. Respir. Crit. Care Med. 154: 237–243
Kankaanranta H, Lindsay MA, Giembycz MA, Zhang X, Moilanen E and Barnes PJ (2000) Delayed eosinophil apoptosis in asthma. J. Allergy Clin. Immunol. 106: 77–83
Matsuda T, Saito H, Fukatsu K, Han I, Inoue T, Furukawa S, Ikeda S and Hidemura A (2001) Cytokine-modulated inhibition of neutrophil apoptosis at local site augments exudative neutrophil functions, and reflects inflammatory response after surgery. Surgery 129: 76–85
Chello M, Mastroroberto P, Quirino A, Cuda G, Perticone F, Cirillo F and Covino E (2002) Inhibition of neutrophil apoptosis after coronary bypass operation with cardiopulmonary bypass. Ann. Thorac. Surg. 73: 123–129
Clancy RM and Abramson SB (1995) Nitric oxide: a novel mediator of inflammation. Proc. Soc. Exp. Biol. Med. 210: 93–101
Heeringa P, van Goor H, Itoh-Lindstrom Y, Maeda N, Falk RJ, Assmann KJ, Kallenberg CG and Jennette JC (2000) Lack of endothelial nitric oxide synthase aggravates murine accelerated anti-glomerular basement membrane glomerulonephritis. Am. J. Pathol. 156: 879–888
Billiar TR, Curran RD, Harbrecht BG, Stuehr DJ, Demetris AJ and Simmons RL (1990) Modulation of nitrogen oxide synthesis in vivo: NG-monomethyl-L- arginine inhibits endotoxin-induced nitrate/nitrate biosynthesis while promoting hepatic damage. J. Leukoc. Biol. 48: 565–569
Veihelmann A, Landes J, Hofbauer A, Dorger M, Refior HJ, Messmer K and Krombach F (2001) Exacerbation of antigen-induced arthritis in inducible nitric oxide synthase-deficient mice. Arthritis Rheum. 44: 1420–1427
McCartney-Francis NL, Song X, Mizel DE and Wahl SM (2001) Selective inhibition of inducible nitric oxide synthase exacerbates erosive joint disease. J. Immunol. 166: 2734–2740
Liu P, Xu B and Hock CE (2001) Inhibition of nitric oxide synthesis by L-name exacerbates acute lung injury induced by hepatic ischemia-reperfusion. Shock 16: 211–217
Ormerod AD, Copeland P, Hay I, Husain A and Ewen SW (1999) The inflammatory, and cytotoxic effects of a nitric oxide releasing cream on normal skin. J. Invest. Dermatol. 113: 392–397
Ross R and Reske-Kunz AB (2001) The role of NO in contact hypersensitivity. Int. Immunopharmacol. 1: 1469–1478
Okuda Y, Sakoda S, Fujimura H and Yanagihara T (1998) Aminoguanidine, a selective inhibitor of the inducible nitric oxide synthase, has different effects on experimental allergic encephalomyelitis in the induction, and progression phase. J. Neuroimmunol. 81: 201–210
Xu LY, Yang JS, Link H, and Xiao BG (2001) SIN-1, a nitric oxide donor, ameliorates experimental allergic encephalomyelitis in Lewis rats in the incipient phase: the importance of the time window. J. Immunol. 166: 5810–5816
Paul-dark MJ, Gilroy DW, Willis D, Willoughby DA and Tomlinson A (2001) Nitric oxide synthase inhibitors have opposite effects on acute inflammation depending on their route of administration. J. Immunol. 166: 1169–1177
Okuda Y, Sakoda S, Fujimura H and Yanagihara T (1997) Nitric oxide via an inducible isoform of nitric oxide synthase is a possible factor to eliminate inflammatory cells from the central nervous system of mice with experimental allergic encephalomyelitis. J. Neuroimmunol. 73: 107–116
Wang BY, Ho HK, Lin PS, Schwarzacher SP, Pollman MJ, Gibbons GH, Tsao PS and Cooke JP (1999) Regression of atherosclerosis: role of nitric oxide, and apoptosis. Circulation 99: 1236–1241
Niebauer J, Schwarzacher SP, Hayase M, Wang B, Kernott RS, Cooke JP and Yeung AC (1999) Local L-arginine delivery after balloon angioplasty reduces monocyte binding, and induces apoptosis. Circulation 100: 1830–1835
Bogdan C (1998) The multiplex function of nitric oxide in (auto) immunity. J. Exp. Med. 187: 1361–1365
Puerta C, Martinez I, Baranda P, Blasco MR, Castejon R, Vargas JA and Garcia-Merino A (2000) Aminoguanidine reduces apoptosis of circulating V Beta 8.2 T lymphocytes in Lewis rats with actively induced experimental autoimmune encephalomyelitis. Association with persistent inflammation of the central nervous system, and lack of recovery. J. Neuroimmunol. 110: 140–150
Ozen S, Usta Y, Sahin-Erdemli I, Orhan D, Gumusel B, Yang B, Gursoy Y, Tulunayo, Dalkara T, Bakkaloglu A and El-Nahas M (2001) Association of nitric oxide production, and apoptosis in a model of experimental nephropathy. Nephrol. Dial. Transplant. 16: 32–38
Rangan GK, Wang Y and Harris DC (2001) Pharmacologic modulators of nitric oxide exacerbate tubulointerstitial inflammation in proteinuric rats. J. Am. Soc. Nephrol. 12: 1696–1705
Shao J, Miyata T, Yamada K, Hanafusa N, Wada T, Gordon KL, Inagi R, Kurokawa K, Fujita T, Johnson RJ and Nangaku M (2001) Protective role of nitric oxide in a model of thrombotic microangiopathy in rats. J. Am. Soc. Nephrol. 12: 2088–2097
Meagher LC, Cousin JM, Seckl JR and Haslett C (1996) Opposing effects of glucocorticoids on the rate of apoptosis in neutrophilic, and eosinophilic granulocytes. J. Immunol. 156: 4422–4428
Galigniana MD, Piwien-Pilipuk G and Assreuy J (1999) Inhibition of glucocorticoid receptor binding by nitric oxide. Mol. Pharmacol. 55: 317–323
Kosonen O, Kankaanranta H, Malo-Ranta U and Moilanen E (1999) Nitric oxide-releasing compounds inhibit neutrophil adhesion to endothelial cells. Eur. J. Pharmacol. 382: 111–117
Li J and Billiar TR (1999) The anti-apoptotic actions of nitric oxide in hepatocytes. Cell Death Differ 6: 952–955
Kolb JP (2000) Mechanisms involved in the pro-, and anti-apoptotic role of NO in human leukemia. Leukemia 14: 1685–1694
Edwards JC, Barry BK, Gruetter DY, Ohlstein EH, Baricos WH and Ignarro LJ (1981) Activation of hepatic guanylate cyclase by nitrosyl-heme complexes. Comparison of unpurified, and partially purified enzyme. Biochem. Pharmacol. 30: 2531–2538
So HS, Park RK, Kim MS, Lee SR, Jung BH, Chung SY, Jun CD and Chung HT (1998) Nitric oxide inhibits c-Jun N-terminal kinase 2 (JNK2) via S-nitrosylation. Biochem. Biophys. Res. Commun. 247: 809–813
Ward C, Chilvers ER, Lawson MF, Pryde JG, Fujihara S, Farrow SN, Haslett C and Rossi AG (1999) NF-kappaB activation is a critical regulator of human granulocyte apoptosis in vitro. J. Biol. Chem. 274: 4309–4318
Watson RW, Rotstein OD, Parodo J, Bitar R, Marshall JC, William R and Watson G (1998) The IL-1 beta-converting enzyme (caspase-1) inhibits apoptosis of inflammatory neutrophils through activation of IL-1 beta. J. Immunol. 161: 957–962
von Knethen A, Callsen D and Brune B (1999) NF-kappaB, and AP-1 activation by nitric oxide attenuated apoptotic cell death in RAW 264.7 macrophages. Mol. Biol. Cell 10: 361–372
Connelly L, Palacios-Callender M, Ameixa C, Moncada S and Hobbs AJ (2001) Biphasic regulation of NF-kappaB activity underlies the pro-, and anti-inflammatory actions of nitric oxide. J. Immunol. 166: 3873–3881
Lander HM, Sehajpal P, Levine DM and Novogrodsky A (1993) Activation of human peripheral blood mononuclear cells by nitric oxide-generating compounds. J. Immunol. 50: 1509–1516
Kalra D, Baumgarten G, Dibbs Z, Seta Y, Sivasubramanian N and Mann DL (2000) Nitric oxide provokes tumor necrosis factor-alpha expression in adult feline myocardium through a cGMP-dependent pathway. Circulation 102: 1302–1307
Zamora R, Alarcon L, Vodovotz Y, Betten B, Kim PK, Gibson KF and Billiar TR (2001) Nitric oxide suppresses the expression of Bcl-2 binding protein BNIP3 in hepatocytes. J. Biol. Chem. 276: 46887–46895
Rossig L, Haendeler J, Hermann C, Malchow P, Urbich C, Zeiher AM and Dimmeler S (2000) Nitric oxide downregulates MKP-3 mRNA levels: involvement in endothelial cell protection from apoptosis. J. Biol. Chem. 275: 25502–25507
Andoh T, Lee SY and Chiueh CC (2000) Preconditioning regulation of bcl-2, and p66shc by human NOSl enhances tolerance to oxidative stress. FASEB J. 14: 2144–2146
Delikouras A, Hayes M, Malde P, Lechler RI and Dorling A (2001) Nitric oxide-mediated expression of Bcl-2, and Bcl-xl, and protection from tumor necrosis factor-alpha-mediated apoptosis in porcine endothelial cells after exposure to low concentrations of xenoreactive natural antibody. Transplantation 71: 599–605
von Knethen A, Callsen D and Brune B (1999) Superoxide attenuates macrophage apoptosis by NF-kappa B, and AP-1 activation that promotes cyclooxygenase-2 expression. J. Immunol. 163: 2858–2866
Rosenberg PA, Li Y, Ali S, Altiok N, Back SA and Volpe JJ (1999) Intracellular redox state determines whether nitric oxide is toxic or protective to rat oligodendrocytes in culture. J. Neurochem. 73: 476–484
Espey MG, Miranda KM, Feelisch M, Fukuto J, Grisham MB, Vitek MP and Wink DA (2000) Mechanisms of cell death governed by the balance between nitrosative and oxidative stress. Ann. N. Y. Acad. Sci. 899: 209–221
Misso NL, Peacock CD, Watkins DN and Thompson PJ (2000) Nitrite generation, and antioxidant effects during neutrophil apoptosis. Free Radic. Biol. Med. 28: 934–943
Melino G, Bernassola F, Knight RA, Corasaniti MT, Nistico G and Finazzi-Agro A (1997) S-nitrosylation regulates apoptosis. Nature 388: 432–433
Mohr S, Zech B, Lapetina EG and Brune B (1997) Inhibition of caspase-3 by S-nitrosation and oxidation caused by nitric oxide. Biochem. Biophys. Res. Commun. 238: 387–391
Zech B, Wilm M, van Eldik R and Brune B (1999) Mass spectrometric analysis of nitric oxide-modified caspase-3. J. Biol. Chem. 274: 20931–20936
Li J, Billiar TR, Talanian RV and Kim YM (1997) Nitric oxide reversibly inhibits seven members of the caspase family via S-nitrosylation. Biochem. Biophys. Res. Commun. 240: 419–424
Rossig L, Fichtlscherer B, Breitschopf K, Haendeler J, Zeiher AM, Mulsch A and Dimmeler S (1999) Nitric oxide inhibits caspase-3 by S-nitrosation in vivo. J. Biol. Chem. 274: 6823–6826
Kim YM, Kim TH, Seol DW, Talanian RV and Billiar TR (1998) Nitric oxide suppression of apoptosis occurs in association with an inhibition of Bcl-2 cleavage, and cytochrome c release. J. Biol. Chem. 273: 31437–31441
Kim YM, Talanian RV and Billiar TR (1997) Nitric oxide inhibits apoptosis by preventing increases in caspase-3-like activity via two distinct mechanisms. J. Biol. Chem. 272: 31138–31148
Dimmeler S, Haendeler J, Nehls M and Zeiher AM (1997) Suppression of apoptosis by nitric oxide via inhibition of interleukin-1 beta-converting enzyme (ICE)-like, and cysteine protease protein (CPP)- 32-like proteases. J. Exp. Med. 185: 601–607
Dimmeler S, Haendeler J, Sause A and Zeiher AM (1998) Nitric oxide inhibits APO-1/Fas-mediated cell death. Cell Growth Differ. 9: 415–422
Torok NJ, Higuchi H, Bronk S and Gores GJ (2002) Nitric oxide inhibits apoptosis downstream of cytochrome c release by nitrosylating caspase 9. Cancer Res. 62: 1648–1653
Williams TE, Ayala A and Chaudry IH (1997) Inducible macrophage apoptosis following sepsis is mediated by cysteine protease activation, and nitric oxide release. J. Surg. Res. 70: 113–118
Knepper-Nicolai B, Savill J and Brown SB (1998) Constitutive apoptosis in human neutrophils requires synergy between calpains, and the proteasome downstream of caspases. J. Biol. Chem. 273: 30530–30536
Clementi E, Brown GC, Feelisch M and Moncada S (1998) Persistent inhibition of cell respiration by nitric oxide: crucial role of S-nitrosylation of mitochondrial complex I, and protective action of glutathione. Proc. Natl. Acad. Sci. USA 95: 7631–7636
Brown GC and Borutaite V (2001) Nitric oxide, mitochondria, and cell death. IUBMB Life 52: 189–195
Sandoval M, Ronzio RA, Muanza DN, Clark DA and Miller MJ (1997) Peroxynitrite-induced apoptosis in epithelial (T84), and macrophage (RAW 264.7) cell lines: effect of legume-derived polyphenols (phytolens). Nitric Oxide 1: 476–483
Wink DA, Grisham MB, Mitchell JB and Ford PC (1996) Direct and indirect effects of nitric oxide in chemical reactions relevant to biology. Methods Enzymol. 268: 12–31
Messmer UK, Ankarcrona M, Nicotera P and Brune B (1994) p53 expression in nitric oxide-induced apoptosis. FEBS Lett. 355: 23–26
Messmer UK and Brune B (1996) Nitric oxide-induced apoptosis: p53-dependent, and p53-independent signalling pathways. Biochem. J. 319(Part 1): 299–305
Raychaudhuri B, Dweik R, Connors MJ, Buhrow L, Malur A, Drazba J, Arroliga AC, Erzurum SC, Kavuru MS and Thomassen MJ (1999) Nitric oxide blocks nuclear factor-kappaB activation in alveolar macrophages. Am. J. Respir. Cell Mol. Biol. 21: 311–316
Welters ID, Menzebach A, Goumon Y, Cadet P, Menges T, Hughes TK, Hempelmann G and Stefano GB (2000) Morphine inhibits NF-kappaB nuclear binding in human neutrophils and monocytes by a nitric oxide-dependent mechanism. Anesthesiology 92: 1677–1684
Fortenberry JD, Owens ML, Chen NX and Brown LA (2001) S-nitrosoglutathione inhibits TNF-alpha-induced NFkappaB activation in neutrophils. Inflamm. Res. 50: 89–95
Matthews JR, Botting CH, Panico M, Morris HR and Hay RT (1996) Inhibition of NF-kappaB DNA binding by nitric oxide. Nucleic Acids Res. 24: 2236–2242
Marshall HE and Stamler JS (2001) Inhibition of NF-kappa B by S-nitrosylation. Biochemistry 40: 1688–1693
Peng HB, Libby P and Liao JK (1995) Induction, and stabilization of I kappa B alpha by nitric oxide mediates inhibition of NF-kappa B. J. Biol. Chem. 270: 14214–14219
Katsuyama K, Shichiri M, Marumo F and Hirata Y (1998) NO inhibits cytokine-induced iNOS expression, and NF-kappaB activation by interfering with phosphorylation, and degradation of IkappaB-alpha. Arterioscler. Thromb. Vasc. Biol. 18: 1796–1802
Glockzin S, von Knethen A, Scheffner M and Brune B (1999) Activation of the cell death program by nitric oxide involves inhibition of the proteasome. J. Biol. Chem. 274: 19581–19586
Tamatani M, Ogawa S, Nunez G and Tohyama M (1998) Growth factors prevent changes in Bcl-2, and Bax expression, and neuronal apoptosis induced by nitric oxide. Cell Death Differ. 5: 911–919
Matsuzaki H, Tamatani M, Mitsuda N, Namikawa K, Kiyama H, Miyake S and Tohyama M (1999) Activation of Akt kinase inhibits apoptosis, and changes in Bcl-2, and Bax expression induced by nitric oxide in primary hippocampal neurons. J. Neurochem. 73: 2037–2046
Ho YS, Liu HL, Duh JS, Chen RJ, Ho WL, Jeng JH, Wang YJ and Lin JK (1999) Induction of apoptosis by S-nitrosoglutathione, and Cu2+ or Ni2+ ion through modulation of bax, bad, and bcl-2 proteins in human colon adenocarcinoma cells. Mol. Carcinogen. 26: 201–211
Jones DR, Broad RM, Comeau LD, Parsons SJ and Mayo MW (2002) Inhibition of nuclear factor kappaB chemosensitizes non-small cell lung cancer through cytochrome c release, and caspase activation. J. Thorac. Cardiovasc. Surg. 123: 310–317
Piantadosi CA, Tatro LG and Whorton AR (2002) Nitric oxide, and differential effects of ATP on mitochondrial permeability transition. Nitric Oxide 6: 45–60
Hortelano S, Dallaporta B, Zamzami N, Hirsch T, Susin SA, Marzo I, Bosca L and Kroemer G (1997) Nitric oxide induces apoptosis via triggering mitochondrial permeability transition. FEBS Lett. 410: 373–377
Stamler JS, Jaraki O, Osborne J, Simon Dl, Keaney J, Vita J, Singel D, Valeri CR and Loscalzo J (1992) Nitric oxide circulates in mammalian plasma primarily as an S-nitroso adduct of serum albumin. Proc. Natl. Acad. Sci. USA 89: 7674–7677
Ramachandran N, Root P, Jiang XM, Hogg PJ and Mutus B (2001) Mechanism of transfer of NO from extracellular S-nitrosothiols into the cytosol by cell-surface protein disulfide isomerase. Proc. Natl. Acad. Sci. USA 98: 9539–9544
Author information
Authors and Affiliations
Corresponding author
Additional information
Edited by G. Melino
Rights and permissions
About this article
Cite this article
Taylor, E., Megson, I., Haslett, C. et al. Nitric oxide: a key regulator of myeloid inflammatory cell apoptosis. Cell Death Differ 10, 418–430 (2003). https://doi.org/10.1038/sj.cdd.4401152
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.cdd.4401152
Keywords
This article is cited by
-
Evaluating the Apoptotic Cell Death Modulatory Activity of Nanoparticles in Men and Women Neutrophils and Eosinophils
Inflammation (2022)
-
Cold bubble humidification of low-flow oxygen does not prevent acute changes in inflammation and oxidative stress at nasal mucosa
Scientific Reports (2021)
-
Effect of bisphenol A on human neutrophils immunophenotype
Scientific Reports (2020)
-
Nitric oxide-generating compound GSNO suppresses porcine circovirus type 2 infection in vitro and in vivo
BMC Veterinary Research (2016)
-
Nitric oxide-mediated apoptosis of neutrophils through caspase-8 and caspase-3-dependent mechanism
Cell Death & Disease (2016)
