
Abstract
Activation-induced cell death (AICD), a process mediated by CD95 and CD95 ligand (CD95L), plays a critical role in regulating homeostasis of the immune system. Although the role of sphingolipids such as ceramides has been suggested to participate in CD95-mediated apoptosis, the exact role of these molecules in this process remains controversial. We employed myriocin, a specific inhibitor of serine palmitoyl-CoA transferase that mediates the first commitment step in sphingolipid synthesis. We found that myriocin could effectively block AICD in T-cell hybridomas and T-cell blasts. However, fumonisin B1, an inhibitor of the final step of ceramide synthesis, or inhibitors of sphingomyelinases did not prevent AICD. Furthermore, ceramide analogues, such as C2 and C6, could not reverse the inhibitory effect of myriocin. Interestingly, sphinganine, an intermediate of ceramide synthesis, completely reversed the inhibitory effect of myriocin, indicating a critical role of sphinganine. Myriocin did not modulate the expression of CD95 or CD95L, instead, it interfered with the early steps of CD95-mediated caspase activation. Therefore, we have uncovered a novel mechanism by which sphingolipid intermediates regulate CD95-mediated apoptosis.
Similar content being viewed by others

Introduction
One of the best-characterized systems in which apoptosis can be demonstrated is activation-induced cell death (AICD) in T cells.1,2 AICD is a fundamental mechanism for the removal of excess peripheral lymphocytes after mounting an immune response.3,4,5,6,7 Defects in this programmed removal mechanism could, therefore, lead to the accumulation of potentially autoreactive lymphocytes. Lymphoproliferation (lpr) and generalized lymphoproliferative disease (gld) are spontaneous mutations in mice that are loss of function mutations of Fas/CD95 and FasL/CD95L, respectively.8 Mice with lpr and gld mutations produce large amounts of autoantibodies and develop an autoimmune disease that resembles human systemic lupus erythematosus.9 Autoimmune lymphoproliferative syndrome (ALPS) pathogenesis in humans has been attributed to defective lymphocyte apoptosis caused by mutations of either the CD95 or CD95L gene.
Studies in the last few years have revealed that CD95 and CD95L play a critical role in AICD. It has been demonstrated that blocking the interaction between CD95 and CD95L could inhibit AICD.10 The unique ‘death domain’ motif in the cytoplasmic region of the CD95 receptor is essential for the initiation of receptor-mediated caspase activation cascade. The binding of CD95L to CD95 induces trimerization, which brings together the intracellular death domains, leading to the recruitment of Fas-associated death domain (FADD).7 The death effector domain in FADD further recruits procaspase 8 (FLICE) to form the death-inducing signaling complex (DISC).11 Upon joining the complex, procaspase 8 is cleaved and becomes activated. Activated caspase 8 initiates the activation of a caspase cascade that includes caspase 3 (CPP32). Caspases can also be activated in response to cellular stress, which often targets mitochondria. Such stimuli result in the release of proapoptotic molecules including apoptosis-inducing factor (AIF) and cytochrome c. Once released into the cytosol, cytochrome c interacts with Apaf-1, dATP/ATP and procaspase 9 to form a complex called the ‘apoptosome’ that can activate procaspase 9.11 Caspase-9 can signal downstream and activate procaspase 3 and procaspase 7. In any case, both pathways lead to the activation of caspase 3. Caspase 3 has been considered as an effector caspase, which then cleaves the inhibitor of caspase-activated deoxyribonuclease (ICAD) to release CAD.12 Once CAD is released, it degrades genomic DNA at the internucleosomal regions and ultimately leads to irreversible apoptosis.
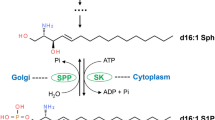
Although the blueprints of the pathways by which CD95 induces apoptosis have been established, the details of the molecular mechanisms are yet to be revealed. One example is the role of sphingolipids in CD95-mediated apoptosis. These lipids are a large class of membrane lipids that have attracted much attention in the recent years because of their diverse roles in biological processes such as cell survival, differentiation and cell death. Sphingolipids are derivatives of the C18 amino alcohols sphingosine and sphinganine. Ceramide is the fundamental structural unit common to all sphingolipids and is one of the most studied members of the sphingolipid family. The attachment of different structures to ceramide via -OH on C-1 of sphingosine forms various types of sphingolipids. The first step in de novo synthesis of ceramide begins with the condensation of palmitoyl CoA with serine to form 3-ketosphinganine, which is catalyzed by serine palmitoyl transferase. 3-Ketosphinganine is subsequently reduced to form sphinganine, then acylated by ceramide synthase to yield dihydroceramide. Finally, dihydroceramide is oxidized to yield ceramide, where a trans-4,5 double bond is introduced by the enzyme dihydroceramide reductase.13 Ceramide may also be produced via the action of several distinct enzymes involved in the cleavage of sphingomyelin (ceramide linked to phosphocholine) by acidic (a) or neutral (n) sphingomyelienases (SMases) (a diagrammatic representation of sphingolipid metabolism is shown in Figure 1). There exist two forms of nSMase: one is cytosolic, Mg2+-independent and the other is membrane-bound, Mg2+-dependent.14,15 Depending on the cell lines and experimental procedures employed, ceramide has been shown to mediate apoptosis,16 cell cycle arrest,17 induction of the JNK and ERK pathway,18 increased cell survival,19 cell proliferation20 and activation of NFκB.21

Diagrammatic representation of the pathways of sphingolipid biosynthesis. The framed lettering shows the target enzymes and their inhibitors
Several studies have indicated a role for ceramide in various types of apoptosis through the activation of aSMase, leading to the modulation of BAD/BAX, Apaf1, cytochrome c and the activation of the SAPK/JNK pathway.13,22 On the other hand, SMases can also be activated by positive signals elicited by PDGF, CD28 and oxidized LDL resulting in the transcription of growth-related genes through the activation of the MAPK pathway and upregulation of a plethora of essential cellular functions like cell growth, differentiation and repair.23,24,25 In the negative signaling pathway, ceramide behaves as a second messenger in the initiation of apoptotic cell death in a variety of pathophysiologic settings.26 Application of exogenous ceramide alone, using cell-permeable ceramide analogues such as C2, C6 and C8 isoforms, is sufficient to initiate apoptosis in some cells.27 In contrast, the use of fumonisin B1, a specific ceramide synthase inhibitor (Figure 1), provides marked protection against chemical hypoxia-induced DNA strand breaks, DNA fragmentation and cell death.28 Recent investigations have shown that in some experimental systems, CD95 signals via ceramide-rich membrane rafts, suggesting that ceramide might participate in the clustering of CD95.29,30
Although many studies have shown the role of ceramide in apoptosis, most of these studies were performed in cell-free systems with analogues of ceramide. In addition, the effect of ceramide has been shown to vary from system to system, with results ranging from regulating apoptosis to promoting mitogenic responses. We have evaluated the importance of ceramide and other sphingolipid intermediates in activation-induced apoptosis, and demonstrated that de novo synthesis of the sphingolipid intermediate, sphinganine, but not ceramide, is important for TCR-signaled cell death following activation. The effect of these intermediates seems to be exerted through modulating the early steps of CD95-mediated caspase activation. This is the first time that sphingolipid intermediates are being shown to be important in the CD95 signaling process.
Results
Myriocin inhibits activation-induced apoptosis
Myriocin (ISP-1, thermozymocidin) is a newly identified immunosuppressant31 with a structure homologous to sphingosine. A natural product of Isaria sinclairii, myriocin specifically inhibits serine palmitoyl transferase,32 the first enzyme in the de novo synthesis of ceramide (Figure 1). Miyake et al.32 found that myriocin suppressed proliferation of an IL-2-dependent mouse cytotoxic T-cell line, CTLL-2, in a dose-dependent manner. Suppression of CTLL-2 proliferation with myriocin was found to be a result of the inhibition of serine palmitoyl transferase (Figure 1). The serine palmitoyl transferase enzyme consists of two subunits, LCB1/SPTLC1 and LCB2/SPTLC2.33 Myriocin inhibits serine palmitoyl transferase activity by binding directly to LCB1 and LCB2.34 Deoxomyriocin is a synthesized analog of myriocin found to be 5 to 10-fold more potent than myriocin (data not shown). Since sphingolipids such as ceramide have been implied to play a critical role in the pathway leading to apoptosis, we first examined the effect of myriocin on AICD. T-cell hybridoma A1.1 cells were activated with anti-CD3 immobilized on tissue culture plastic plates overnight in the presence or absence of myriocin. We found that myriocin inhibited AICD in a concentration-dependent manner, with 50% inhibition at 3.125 μM and complete inhibition at 6.25 μM. As shown in Figure 2a, A1.1 cells activated with anti-CD3 show membrane blebbing, characteristic of cells undergoing apoptosis. On the other hand, A1.1 cells activated with anti-CD3 that were also treated with 6.25 μM myriocin appeared round and healthy, illustrating that myriocin is effective in inhibiting AICD. At this concentration, myriocin did not have any effect on cell proliferation in unactivated cells (data not shown). We also looked at whether deoxomyriocin could inhibit AICD. Anti-CD3 treatment induced apoptosis in A1.1 cells as demonstrated by the presence of a hypodiploid peak in DNA content analysis (Figure 2b). Anti-CD3-treated A1.1 cells also treated with deoxomyriocin did not have a hypodiploid peak, indicating that deoxomyriocin can also inhibit apoptosis in A1.1 cells.

Myriocin inhibits AICD. (a) A1.1 cells were activated with plastic-bound anti-CD3 alone with or without 6.25 μM myriocin. Photographs were taken 14 h after activation. (b) and (c) A1.1 cells were activated with anti-CD3 in the presence and absence of 6.25 μM deoxomyriocin or fumonisin B1 for 14 h. Apoptosis was determined by DNA content analysis. The marked region represents percent of apoptotic cells in the subdiploid peak
To reveal whether de novo synthesis of ceramide plays a role in the protection of cells from activation-induced apoptosis, we examined the effect of the ceramide synthase inhibitor, Fumonisin B1 (FB1),35 which inhibits the final step in ceramide synthesis. FB1 is the most common mycotoxin produced by Fusarium moniliforme. The structure of FB1 resembles sphingosine and studies indicate that FB1 inhibits sphingolipid biosynthesis by inhibiting ceramide synthase activity. FB1 interacts with the binding sites for sphinganine and fatty acyl-coenzyme A (CoA)36 in a competitive manner, causing sphinganine to accumulate. Wang et al.35 showed that the IC50 of FB1 was approximately 0.1 μM for inhibiting the incorporation of serine into sphingosine. Anti-CD3 treatment induced apoptosis in A1.1 cells as demonstrated by the hypodiploid peak in DNA content analysis (Figure 2c). Activated A1.1 cells treated with FB1 also had a hypodiploid peak. Therefore, blockade of ceramide synthesis using FB1 does not prevent AICD. We also found that ceramide analogues C2 and C6 could not induce apoptosis in A1.1 cells, nor could these ceramide analogues reverse the inhibitory effect of myriocin upon anti-CD3 activation (data not shown). These results reveal that although sphingolipid synthesis is required as indicated by the capability of myriocin to inhibit AICD, de novo synthesis of ceramide is not necessary for activation-induced apoptosis.
To test the effect of myriocin on activation-induced apoptosis in other cells, we employed another T-cell hybridoma IE5. As in A1.1 cells, AICD in IE5 cells is also dependent on Fas and FasL interaction (data not shown). As shown in Figure 3a, anti-CD3-induced AICD in these cells is blocked by myriocin. In addition, we also generated T-cell blasts from freshly isolated splenocytes and tested for AICD according to the established protocol.3,4 We found that AICD in T-cell blasts was also inhibited by myriocin (Figure 3b). Furthermore, we have found that apoptosis in A1.1 cells induced by dexamethasone and cisplatin is not affected by myriocin (data not shown), indicating the specificity of myriocin.

Myriocin inhibits AICD in other cells. T-cell hybridoma IE5 cells (a) and T cell blasts (b) were activated with plastic-bound anti-CD3 for 12 h in the presence or absence of 6.25 μM myriocin. Photographs represent cellular morphology, and percentage on the top left corners shows the ratio of apoptotic cells as determined by DNA content analysis
Sphinganine reverses the effect of myriocin
The synthesis of sphingolipids is a multistep process. Sphinganine is an intermediate upstream of ceramide synthase, an enzyme sensitive to FB1. If this intermediate participates in Fas-mediated apoptosis, it should be able to reverse the inhibitory effect of myriocin. To test this hypothesis, we activated A1.1 cells with anti-CD3 in the presence of both myriocin and sphinganine, and assessed apoptosis by DNA content analysis. As shown above, myriocin can inhibit AICD, as seen by the absence of the subdiploid peak. Interestingly, when cells were treated with both myriocin and sphinganine, a subdiploid peak reappeared (Figure 4), indicating that cells underwent apoptosis. This suggests that sphinganine is an important intermediate of the sphingolipid synthesis pathway participating in Fas-mediated apoptosis.

Sphinganine reverses the effect of myriocin. A1.1 cells were activated with anti-CD3 in the presence and absence of 6.25 μM deoxomyriocin and 3.13 μM sphinganine for 14 h. Apoptosis was determined by DNA content analysis. The marked region represents percent of apoptotic cells in the subdiploid peak
Myriocin does not inhibit IL-2 production
Activation of A1.1 T-cell hybridoma through the T-cell receptor by antigen plus MHC or with anti-CD3 leads to apoptosis.37 Activation of T-cell hybridomas also leads to the production of cytokines such as IL-2.38 In order to determine whether myriocin inhibits AICD by interfering with TCR signaling, we examined if myriocin had an effect on IL-2 production. A1.1 cells were treated with myriocin, C2 or C6 ceramides in the presence or absence of anti-CD3 for 14 h. Culture supernatants were examined for IL-2 production by ELISA. As shown in Figure 5, unactivated A1.1 cells do not produce detectable amounts of IL-2, while anti-CD3 activation leads to a significant increase in IL-2 production. Cells treated with anti-CD3 as well as myriocin also produced significant amounts of IL-2, illustrating that TCR signaling is not compromised and that myriocin inhibits apoptosis downstream of TCR signaling. Similarly, FB1, C2, C6 and sphinganine also did not affect IL-2 production. Therefore, sphingolipids are not involved in general TCR activation process.

Myriocin does not inhibit activation-induced IL-2 production. A1.1 cells were treated with 6.25 μM myriocin, FB1, sphinganine, C2 or C6 ceramide in the presence or absence of anti-CD3 for 14 h. Culture supernatants were examined for IL-2 production by ELISA
Myriocin does not affect CD95 or CD95L expression
Crosslinking the TCR of T-cell hybridomas leads to AICD. AICD in T-cell hybridomas have been shown to require the expression of CD95 and CD95L.39 We tested whether the inhibitory effect of myriocin on AICD is because of its effect on activation-induced CD95 and CD95L expression. A1.1 cells express low basal levels of CD95 that increase within 2–4 h upon activation with anti-CD3. On the other hand, A1.1 cells do not express CD95L unless they are activated with anti-CD3.40 CD95L expression can be seen in A1.1 cells at 4–6 h after activation with anti-CD3.41 We found that neither myriocin nor FB1 affects CD95 or CD95L expression upon activation with ani-CD3 (Figure 6). Therefore, the effect of sphingolipids is not exerted by interfering with activation-induced CD95 and CD95L expression.

Myriocin does not affect CD95 and CD95L expression. A1.1 cells were treated with or without anti-CD3 in the presence or absence of 6.25 μM myriocin, fumonisin B1 or sphinganine for 4 h. Total RNA was isolated for Northern blot analysis for the expression of CD95, CD95L and GAPDH
Myriocin directly inhibits CD95 signaling
Since myriocin does not affect CD95 and CD95L expression, its effect is likely through the death signal after Fas stimulation. Therefore, we tested the effect of myriocin on CD95-mediated cell death signaling. DNA content analysis of A1.1 cells shows that few cells are in the subdiploid peak (Figure 7a). Treatment with anti-CD3 dramatically increased the number of cells in the subdiploid peak. As demonstrated previously,2 cells treated with both anti-CD3 and CsA are inhibited from undergoing apoptosis, illustrated by the diminished subdiploid peak. CsA inhibits FasL expression, thereby inhibiting apoptosis.42 However, cells rescued by CsA are still sensitive to Fas ligation with JO2. JO2 is an anti-mouse Fas monoclonal antibody with activation capacity. Interestingly, cells rescued with CsA and further treated with JO2 are still inhibited from undergoing apoptosis by myriocin, indicating that the effect of myriocin is downstream of the Fas death signal. In order to further support that myriocin affects CD95-mediated death pathway, we utilized A20 B cells. A20 is a murine B-cell line that expresses CD95 and is very sensitive to CD95 ligation-induced apoptosis. Cells were incubated with JO2 in the presence or absence of myriocin. We found that JO2-induced apoptosis also was inhibited by myriocin in A20 B cells (Figure 7b). Since myriocin does not modulate the transcriptional regulation of CD95 or CD95L, this result suggests that the inhibitory effect of myriocin is specifically involved in the CD95-death pathway.

Myriocin directly inhibits CD95 signaling. (a) A1.1 cells were treated with anti-CD3, 200 ng/ml cyclosporin A, 200 ng/ml JO2 in the presence or absence of 6 μM myriocin for 12 h. Apoptosis was determined by DNA content analysis. The marked region represents percent of apoptotic cells in the subdiploid peak. (b) A20 cells were treated with 10 ng/ml JO2 in the presence or absence of 6 μM myriocin for 12 h. Apoptosis was determined by DNA content analysis
Myriocin inhibits caspases 3, 8 and 9 activation
CD95 is a cell surface death receptor that belongs to the tumor necrosis factor receptor superfamily. Upon binding of CD95 to CD95L, CD95 trimerizes and brings together three intracellular death domains that bind to the death domain of FADD. FADD also contains a death effector domain (DED) that recruits the DED of procaspase 8 to form the death-inducing signaling complex (DISC). When procaspase 8 is bound to the DISC, it is cleaved in order to yield active caspase 8, thereby initiating the caspase cascade.43 Since myriocin did not affect CD95 or CD95L expression, we examined whether it affects the death signaling pathway. Figure 8 illustrates that A1.1 cells treated with anti-CD3 have high levels of caspases 3 (Figure 8a), 8 (Figure 8b) and 9 (Figure 8c) activity. The activation of these caspases was inhibited by myriocin. Since both caspases 8 and 9, two initiators of the caspase cascade, were inhibited, it is likely that the effect of myriocin is exerted through modulating the DISC formation process.

Myriocin inhibits caspases -3, -8 and -9 activation. A1.1 cells were activated with anti-CD3 and treated with 12.5 μM myriocin for 5 h. Cells were harvested, lysed and analyzed for caspase activity using Apoalert Caspase-3 (a), caspase-8 (b) and caspase-9 (c) fluorescent assay kit. Fluorescence was determined at 400 nm excitation and 505 nm emission
Discussion
Sphingolipids are key components of the lipid bilayer that can also serve as vital second messengers in cell survival, proliferation, cell–cell communication, cytokine production and apoptosis.44,45,46 The role of sphingolipids in T lymphocytes following TCR activation is beginning to be identified. For example, a recent study has revealed that membrane rafts play a critical role in concentrating MHC class II molecules into microdomains that allow efficient antigen presentation at low ligand densities.47 The importance of modular interaction during transmembrane signaling is rapidly being realized. These membrane microdomains or rafts serve as privileged sites where receptors and proximal signaling molecules optimally interact.48 However, the role of membrane rafts in AICD is currently not known. In this study, we addressed the possible involvement of sphingolipids in the regulation of AICD. We utilized two enzyme inhibitors and determined whether it is ceramide or another sphingolipid intermediate that is important for AICD in A1.1 T cells. A1.1 cells were analyzed following activation with anti-CD3 and treatments with sphingolipid intermediates or sphingolipid synthesis inhibitors, and found an important role of the sphingolipid synthesis intermediates in the modulation of the CD95 death pathway. Our results indicate that the effect of the sphingolipid intermediates is associated with the early steps of CD95-mediated caspase activation, likely by participating in DISC formation.
Sphingolipid synthesis starts with the condensation of palmitoyl CoA with serine to form 3-ketosphinganine, a process catalyzed by serine palmitoyl transferase. 3-Ketosphinganine is subsequently reduced to form sphinganine, then acylated by ceramide synthase to yield dihydroceramide, which is further oxidized to yield ceramide, where a trans-4,5 double bond is introduced by the enzyme dihydroceramide reductase.13 In this study, we employed a serine palmitoyl-CoA transferase inhibitor, myriocin, to block sphingolipid synthesis. This inhibitor has been shown to be highly effective and specific in inhibiting the activity of serine-palmitoyl transferase by binding to the enzyme.32,34 As previously mentioned, serine-palmitoyl transferase consists of two subunits: LCB1 and LCB2. Myriocin specifically inhibits serine-palmitoyl transferase activity by binding to LCB1 and LCB2.34 We have discovered that inhibition of serine-palmitoyl transferase with myriocin, but not of ceramide synthase with FB1, could abrogate CD95-mediated apoptosis. Since myriocin was able to inhibit apoptosis and FB1 was not, it is conceivable that a sphingolipid intermediate upstream of ceramide synthase and downstream of serine-palmitoyl transferase, rather than ceramide itself, is critical for CD95-induced activation of the caspase cascade. Consistent with this notion, we found that addition of sphinganine, but not ceramide analogues, could reverse the inhibitory effect of myriocin.
Initial studies on the effects of myriocin revealed that it inhibits both CTLL-2 cell proliferation and mouse allogeneic mixed lymphocyte reaction by inhibiting serine-palmitoyl transferase.31,32 It was later discovered that the decrease in CTLL-2 cell number upon treatment with myriocin was not because of the inhibition of cell cycle progression but a consequence of apoptosis.49 However, the same group found that myriocin did not induce apoptosis in a mouse pro-B cell line, indicating that the effect of myriocin depends on the cell type. Our experiments show that myriocin does not affect IL-2 production (Figure 5) and is similar to the results from Fujita et al.31 Inhibition of apoptosis by myriocin is not limited to A1.1 cells or to CD95-mediated apoptosis. As we have shown, myriocin also inhibits apoptosis in A20 B cells (Figure 7b), IE5 T-cell hybridoma (Figure 3a) and freshly generated T-cell blasts (Figure 3b). However, myriocin does not seem to affect dexamethasone-induced apoptosis of thymocytes (data not shown). The various effects of myriocin are probably owing to the different amounts and type of sphingolipids expressed in different cell types. Ratios of different sphingolipid intermediates might determine what effect myriocin will have on a specific cell type.
Toxicological studies with FB1 reveal that exposure to this toxin leads to an accumulation of high concentrations of the cytotoxic sphingolipid sphinganine.50,51 By inhibiting ceramide synthesis with FB1, sphinganine is prevented from being converted to ceramide, thus, sphinganine accumulates and facilitates cell death. It is worthy of note that although we did not see an inhibitory effect of FB1 on activation-induced apoptosis, we found that FB1 could inhibit radiation-induced apoptosis in A1.1 cells (data not shown). Therefore, the amount of FB1 we used is effective in modulating ceramide production and the effect we saw on AICD is not because of a general toxicity.
Our investigation has excluded the possibility that myriocin inhibits AICD by modulating CD95 and CD95L expression. We could not determine whether CD95 or CD95L surface expression was affected by myriocin because available antibodies do not stain FasL on A1.1 cells. However, in other T-cell hybridoma IE5, FasL could be detected by surface staining. We have found that activation-induced CD95 and CD95L expression on IE5 surface is not affected by myriocin (data not shown). Using Northern blot analysis, we discovered that CD95 and CD95L expressions were not affected by myriocin; therefore, we examined the effect of myriocin on CD95-induced cell death process. The ability of myriocin to prevent CD95-triggered apoptosis was supported by data from A20 cells. A20 cells treated with JO2 undergo CD95-mediated apoptosis. However, A20 cells treated with both JO2 and myriocin did not undergo apoptosis. This also suggests that myriocin inhibits the cell death signal following CD95 ligation. Since myriocin seemed to be inhibiting apoptosis following CD95 ligation, we examined whether caspase activation was affected. We found that myriocin is able to inhibit caspases 3, 8 and 9 activation. These results indicate modulation of caspases by sphingolipids during AICD, pointing to a novel regulatory mechanism. Thus, our studies reveal the requirement of de novo sphingolipid synthesis in AICD. Exogenous administration of sphinganine has been shown to induce apoptosis in hepatoma cells, a mechanism which involves caspase activation.52 Furthermore, CD95-mediated cell death has been shown previously to involve sphingolipids,53,54,55 but in a ceramide-independent mechanism.56,57 The precise mechanism of sphinganine-modulated caspase activation is not known; however, it may be involved with the formation of the DISC, since caspase 8 activation requires its recruitment into the DISC. Therefore, our observation that caspase activation is inhibited by myriocin may be because of the lack of DISC formation. We are currently determining whether myriocin can inhibit DISC formation and investigating whether raft formation is required.
Materials and Methods
Cells and reagents
A1. 1 murine T-cell hybridoma cells were maintained in RPMI-1640 media (GIBCO, Gaithersburg, MD, USA) with 5% FBS (GIBCO, Gaithersburg, MD, USA) and full complement of antibiotics/antimycotics in humidified atmosphere supplemented with 5% CO2 at 37°C. A20 cells were maintained as described by Mueller and Scott.58 Myriocin was obtained from Dr. Fujita. Fumonisin B1 (FB1), C2 Ceramide and C6 Ceramide, were purchased from Calbiochem (La Jolla, CA, USA). Sphinganine (DL-erythro-dihydro-sphingosine) was purchased from Sigma (St. Louis, MO, USA).
Generation of T-cell blasts
Murine splenocytes were isolated from 10-week-old female Balb/c mice (National Cancer Institute, Frederick, MD, USA). Single-cell suspension was generated from spleen by pressing between the frosted ends of microscope slides and passing through 70 μm nylon cell strainers. Cells were activated with anti-CD3 for 48 h and then maintained in the presence of IL-2 for another 48 h. Cells were maintained in RPMI 1640 medium (Gibco/BRL, Gaithersburg, MD, USA), supplemented with 2 mM L-glutamine, 50 mM 2-mercaptoethanol, 10% heat-inactivated fetal bovine serum (Sigma, St. Louis, MO, USA), and 10 mM gentamycin.
DNA content analysis
After treatment, cells were fixed with 70% ethanol for at least 30 min at 4°C, followed by two washes with PBS. The fixed splenocytes were then incubated in PBS containing propidium iodide (Sigma, St. Louis, MO, USA) at 50 μg/ml and RNase (Boehringer Mannheim, Indianapolis, IN, USA) at 0.1 mg/ml at room temperature for 30 min. DNA content was determined by flow cytometry on FacScan (Becton Dickinson, San Jose, CA, USA). The FL2 intensity was plotted as histograms on a linear scale. Apoptotic cells were shown as a hypodiploid peak.
Northern blotting
Total RNA was isolated with affinity columns (QIAGEN, Chatsworth, CA, USA), according to the manufacturer's recommended protocol. RNA samples were fractionated on 1% agarose/2.2 M formaldehyde denaturing gel, and transferred onto a Nytran membrane (Schleicher & Schuell, Inc., Keene, NH, USA). Mouse CD95 and CD95L cDNA probes (from Dr. Shigekazu Nagata, Osaka Bioscience Institute, Japan) were randomly primed with [32P]-dCTP (Boehringer Mannheim, Indianapolis, IN, USA) according to the manufacturer's instructions. Prehybridization and hybridization were carried out at 42°C in a solution containing 5 × SSC (10 × SSC is 1.5 M NaCl, 0.15 M sodium citrate), 2.5 mM EDTA, 0.1% SDS, 5 × Denhardt's solution, 2 mM sodium pyrophosphate, 50 mM sodium phosphate, and 50% formamide. Membranes were washed with 0.2 × SSC, 0.1% SDS at 56°C for 1 h and hybridization signals were detected by autoradiography.
IL-2 ELISA
IL-2 in culture supernatants was detected with the cytoscreen Immunoassay kit (Biosource, Camarillo, CA, USA) according to the manufacturer's recommended protocol. Recombinant murine IL-2 was used as standard. Briefly, the tissue culture supernatant was diluted in a standard diluent buffer included in the kit. Standard cytokines and samples were incubated on the first respective cytokine-specific antibodies-coated microtiter plates for 1.5 h at 37°C. After washing, the bound cytokines were detected with biotinylated second cytokine-specific antibody and streptavidin peroxidase. The amount of cytokines was determined by the addition of tetramethyl benzidine. The amount of IL-2 was determined by recession curves obtained from recombinant murine IL-2 provided in the kit.
Caspase assay
Caspases-8, -9 and -3 activities were determined using ApoAlert Caspase Fluorescent Assay Kits (Clontech, Palo Alto, CA, USA) according to the manufacturer's recommended protocol. Briefly, upon treatments, 2 × 106 cells were resuspended in 50 μl of lysis buffer included in the kits and incubated on ice for 10 min. Upon centrifugation, the lysate supernatant was isolated and mixed with a 2 × reaction buffer/DTT mix. Upon addition of the specific substrate peptide, labeled with chromophore p-nitroaniline, the reaction was carried out at 37°C for 1 h. The activity of caspases was determined at 400 nm excitation and 505 nm emission.
Abbreviations
- AICD:
-
activation-induced cell death
- Lpr:
-
lymphoproliferation
- gld:
-
generalized lymphoproliferative disease
- ALPS:
-
autoimmune lymphoproliferative syndrome
- DISC:
-
death-inducing signaling complex
References
Mercep M, Noguchi PD and Ashwell JD (1989) The cell cycle block and lysis of an activated T cell hybridoma are distinct processes with different Ca2+ requirements and sensitivity to cyclosporine A. J. Immunol. 142: 4085–4092
Shi YF, Sahai BM and Green DR (1989) Cyclosporin A inhibits activation-induced cell death in T-cell hybridomas and thymocytes. Nature 339: 625–626
Lenardo MJ (1991) Interleukin-2 programs mouse alpha beta T lymphocytes for apoptosis. Nature 353: 858–861
Radvanyi LG, Mills GB and Miller RG (1993) Religation of the T cell receptor after primary activation of mature T cells inhibits proliferation and induces apoptotic cell death. J. Immunol. 150: 5704–5715
Russell JH (1995) Activation-induced death of mature T cells in the regulation of immune responses. Curr. Opin. Immunol. 7: 382–388
Scott DW, Grdina T and Shi Y (1996) T cells commit suicide, but B cells are murdered! J. Immunol. 156: 2352–2356
Sharma K, Wang RX, Zhang LY, Yin DL, Luo XY, Solomon JC, Jiang RF, Markos K, Davidson W, Scott DW et al. (2000) Death the Fas way: regulation and pathophysiology of CD95 and its ligand. Pharmacol. Ther. 88: 333–347
Nagata S and Suda T (1995) Fas and Fas ligand: lpr and gld mutations. Immunol. Today 16: 39–43
Cohen PL and Eisenberg RA (1991) Lpr and gld: single gene models of systemic autoimmunity and lymphoproliferative disease. Annu. Rev. Immunol. 9: 243–269
Nagata S and Golstein P (1995) The Fas death factor. Science 267: 1449–1456
Bratton SB, MacFarlane M, Cain K and Cohen GM (2000) Protein complexes activate distinct caspase cascades in death receptor and stress-induced apoptosis. Exp. Cell Res. 256: 27–33
Tang D and Kidd VJ (1998) Cleavage of DFF-45/ICAD by multiple caspases is essential for its function during apoptosis. J. Biol. Chem. 273: 28 549–28 552
Bose R, Verheij M, Haimovitz-Friedman A, Scotto K, Fuks Z and Kolesnick R (1995) Ceramide synthase mediates daunorubicin-induced apoptosis: an alternative mechanism for generating death signals. Cell 82: 405–414
Okazaki T, Bielawska A, Domae N, Bell RM and Hannun YA (1994) Characteristics and partial purification of a novel cytosolic, magnesium-independent, neutral sphingomyelinase activated in the early signal transduction of 1 alpha,25-dihydroxyvitamin D3-induced HL-60 cell differentiation. J. Biol. Chem. 269: 4070–4077
Spence MW, Wakkary J, Clarke JT and Cook HW (1982) Localization of neutral magnesium-stimulated sphingomyelinase in plasma membrane of cultured neuroblastoma cells. Biochim. Biophys. Acta 719: 162–164
Hannun YA and Obeid LM (1995) Ceramide: an intracellular signal for apoptosis. Trends Biochem. Sci. 20: 73–77
Jayadev S, Liu B, Bielawska AE, Lee JY, Nazaire F, Pushkareva M, Obeid LM and Hannun YA (1995) Role for ceramide in cell cycle arrest. J. Biol. Chem. 270: 2047–2052
Pena LA, Fuks Z and Kolesnick R (1997) Stress-induced apoptosis and the sphingomyelin pathway. Biochem. Pharmacol. 53: 615–621.
Ping SE and Barrett GL (1998) Ceramide can induce cell death in sensory neurons, whereas ceramide analogues and sphingosine promote survival. J. Neurosci. Res. 54: 206–213
Olivera A, Buckley NE and Spiegel S (1992) Sphingomyelinase and cell-permeable ceramide analogs stimulate cellular proliferation in quiescent Swiss 3T3 fibroblasts. J. Biol. Chem. 267: 26 121–26 127
Boland MP and O'Neill LA (1998) Ceramide activates NFkappaB by inducing the processing of p105. J. Biol. Chem. 273: 15 494–15 500
Kolesnick RN and Kronke M (1998) Regulation of ceramide production and apoptosis. Annu. Rev. Physiol. 60: 643–665
Jarvis WD and Grant S (1998) The role of ceramide in the cellular response to cytotoxic agents. Curr. Opin. Oncol. 10: 552–559.
Spiegel S and Merrill Jr AH (1996) Sphingolipid metabolism and cell growth regulation. FASEB J. 10: 1388–1397
Spiegel S, Foster D and Kolesnick R (1996) Signal transduction through lipid second messengers. Curr. Opin. Cell Biol. 8: 159–167
Adam-Klages S, Schwandner R, Adam D, Kreder D, Bernardo K and Kronke M (1998) Distinct adapter proteins mediate acid versus neutral sphingomyelinase activation through the p55 receptor for tumor necrosis factor. J. Leukoc. Biol. 63: 678–682
Irie F and Hirabayashi Y (1998) Application of exogenous ceramide to cultured rat spinal motoneurons promotes survival or death by regulation of apoptosis depending on its concentrations. J. Neurosci. Res. 54: 475–485
Ueda N, Kaushal GP, Hong X and Shah SV (1998) Role of enhanced ceramide generation in DNA damage and cell death in chemical hypoxic injury to LLC-PK1 cells. Kidney Int. 54: 399–406
Grassme H, Schwarz H and Gulbins E (2001) Molecular mechanisms of ceramide-mediated CD95 clustering. Biochem. Biophys. Res. Commun. 284: 1016–1030
Grassme H, Jekle A, Riehle A, Schwarz H, Berger J, Sandhoff K, Kolesnick R and Gulbins E (2001) CD95 signaling via ceramide-rich membrane rafts. J. Biol. Chem. 276: 20 589–20 596
Fujita T, Inoue K, Yamamoto S, Ikumoto T, Sasaki S, Toyama R, Chiba K, Hoshino Y and Okumoto T (1994) Fungal metabolites. Part 11. A potent immunosuppressive activity found in Isaria sinclairii metabolite. J. Antibiot. (Tokyo) 47: 208–215
Miyake Y, Kozutsumi Y, Nakamura S, Fujita T and Kawasaki T (1995) Serine palmitoyltransferase is the primary target of a sphingosine-like immunosuppressant, ISP-1/myriocin. Biochem. Biophys. Res. Commun. 211: 396–403
Hanada K, Hara T and Nishijima M (2000) Purification of the serine palmitoyltransferase complex responsible for sphingoid base synthesis by using affinity peptide chromatography techniques. J. Biol. Chem. 275: 8409–8415
Chen JK, Lane WS and Schreiber SL (1999) The identification of myriocin-binding proteins. Chem. Biol. 6: 221–235
Wang E, Norred WP, Bacon CW, Riley RT and Merrill Jr AH (1991) Inhibition of sphingolipid biosynthesis by fumonisins. Implications for diseases associated with Fusarium moniliforme. J. Biol. Chem. 266: 14 486–14 490
Merrill Jr AH, van Echten G, Wang E and Sandhoff K (1993) Fumonisin B1 inhibits sphingosine (sphinganine) N-acyltransferase and de novo sphingolipid biosynthesis in cultured neurons in situ. J. Biol. Chem. 268: 27 299–27 306
Shi YF, Szalay MG, Paskar L, Sahai BM, Boyer M, Singh B and Green DR (1990) Activation-induced cell death in T cell hybridomas is due to apoptosis. Morphologic aspects and DNA fragmentation. J. Immunol. 144: 3326–3333
Fotedar A, Boyer M, Smart W, Widtman J, Fraga E and Singh B (1985) Fine specificity of antigen recognition by T cell hybridoma clones specific for poly-18: a synthetic polypeptide antigen of defined sequence and conformation. J. Immunol. 135: 3028–3033
Brunner T, Mogil RJ, LaFace D, Yoo NJ, Mahboubi A, Echeverri F, Martin SJ, Force WR, Lynch DH, Ware CF et al. (1995) Cell-autonomous Fas (CD95)/Fas-ligand interaction mediates activation-induced apoptosis in T-cell hybridomas. Nature 373: 441–444
Wang R, Zhang L, Yin D, Mufson RA and Shi Y (1998). Protein kinase C regulates Fas (CD95/APO-1) expression. J. Immunol. 161: 2201–2207
Wang R, Zhang L, Zhang X, Moreno J, Luo X, Tondravi M and Shi Y (2001) Differential regulation of the expression of CD95 ligand, receptor activator of nuclear factor-kappa B ligand (RANKL), TNF-related apoptosis-inducing ligand (TRAIL), and TNF-alpha during T cell activation. J. Immunol. 166: 1983–1990
Brunner T, Yoo NJ, LaFace D, Ware CF and Green DR (1996) Activation-induced cell death in murine T cell hybridomas. Differential regulation of Fas (CD95) versus Fas ligand expression by cyclosporin A and FK506. Int. Immunol. 8: 1017–1026. abs.html
Budihardjo I, Oliver H, Lutter M, Luo X and Wang X (1999) Biochemical pathways of caspase activation during apoptosis. Annu. Rev. Cell Dev. Biol. 15: 269–290
Merrill Jr AH, Sullards MC, Wang E, Voss KA and Riley RT (2001) Sphingolipid metabolism: roles in signal transduction and disruption by fumonisins. Environ Health Perspect. 109(Suppl. 2): 283–289
Flores I, Jones DR and Merida I (2000) Changes in the balance between mitogenic and antimitogenic lipid second messengers during proliferation, cell arrest, and apoptosis in T-lymphocytes. FASEB J. 14: 1873–1875
Ballou LR (1992) Sphingolipids and cell function. Immunol. Today 13: 339–341
Anderson HA, Hiltbold EM and Roche PA (2000) Concentration of MHC class II molecules in lipid rafts facilitates antigen presentation. Nat. Immunol. 1: 156–162
Hoessli DC, Ilangumaran S, Soltermann A, Robinson PJ, Borisch B and Nasir UdD (2000) Signaling through sphingolipid microdomains of the plasma membrane: the concept of signaling platform. Glycoconj. J. 17: 191–197
Nakamura S, Kozutsumi Y, Sun Y, Miyake Y, Fujita T and Kawasaki T (1996) Dual roles of sphingolipids in signaling of the escape from and onset of apoptosis in a mouse cytotoxic T-cell line, CTLL-2. J. Biol. Chem. 271: 1255–1257
Humpf HU, Schmelz EM, Meredith FI, Vesper H, Vales TR, Wang E, Menaldino DS, Liotta DC and Merrill Jr AH (1998). Acylation of naturally occurring and synthetic 1-deoxysphinganines by ceramide synthase. Formation of N-palmitoyl-aminopentol produces a toxic metabolite of hydrolyzed fumonisin, AP1, and a new category of ceramide synthase inhibitor. J. Biol. Chem. 273: 19 060–19 064
Schmelz EM, Dombrink-Kurtzman MA, Roberts PC, Kozutsumi Y, Kawasaki T and Merrill Jr AH (1998) Induction of apoptosis by fumonisin B1 in HT29 cells is mediated by the accumulation of endogenous free sphingoid bases. Toxicol. Appl. Pharmacol. 148: 252–260
Hung WC, Chang HC and Chuang LY (1999) Activation of caspase-3-like proteases in apoptosis induced by sphingosine and other long-chain bases in Hep3B hepatoma cells. Biochem. J. 338: 161–166
Brenner B, Ferlinz K, Grassme H, Weller M, Koppenhoefer U, Dichgans J, Sandhoff K, Lang F and Gulbins E (1998) Fas/CD95/Apo-I activates the acidic sphingomyelinase via caspases. Cell Death Differ. 5: 29–37
De Maria R, Rippo MR, Schuchman EH and Testi R (1998). Acidic sphingomyelinase (ASM) is necessary for fas-induced GD3 ganglioside accumulation and efficient apoptosis of lymphoid cells. J. Exp. Med. 187: 897–902
Tepper CG, Jayadev S, Liu B, Bielawska A, Wolff R, Yonehara S, Hannun YA and Seldin MF (1995) Role for ceramide as an endogenous mediator of Fas-induced cytotoxicity. Proc. Natl. Acad. Sci. USA 92: 8443–8447
Hsu SC, Wu CC, Luh TY, Chou CK, Han SH and Lai MZ (1998) Apoptotic signal of Fas is not mediated by ceramide. Blood 91: 2658–2663
Watts JD, Gu M, Polverino AJ, Patterson SD and Aebersold R (1997) Fas-induced apoptosis of T cells occurs independently of ceramide generation. Proc. Natl. Acad. Sci. USA 94: 7292–7296
Mueller CM and Scott DW (2000) Distinct molecular mechanisms of Fas resistance in murine B lymphoma cells. J. Immunol. 165: 1854–1862
Acknowledgements
We thank Dr. Liying Zhang, Miss Jennifer Hinshaw, Dr. Xiaoren Zhang, Dr. Ruoxiang Wang, Dr. Achsah Keegan, Dr. David Scott and Dr. Stephan Ladisch for their discussions and technical help. This work was supported by National Institutes of Health Grants R01AI43384, R01AI50222 and R01CA76492 to YFS and R01CA76492-03SI to JCS. KS is a recipient of Fonds de la recherche en Sante du Quebec postdoctoral fellowship. This work will be submitted in partial fulfillment of the requirements for the Doctor of Philosophy degree (J.C.S.), Molecular and Cellular Oncology Program, Institute for Biomedical Sciences of the Columbian School of Arts and Sciences, The George Washington University.
Author information
Authors and Affiliations
Corresponding author
Additional information
Edited by DR Green
Rights and permissions
About this article
Cite this article
Solomon, J., Sharma, K., Wei, L. et al. A novel role for sphingolipid intermediates in activation-induced cell death in T cells. Cell Death Differ 10, 193–202 (2003). https://doi.org/10.1038/sj.cdd.4401136
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.cdd.4401136
