
Abstract
Caspase activation may occur in a direct fashion as a result of CD95 death receptor crosslinking (exogenous pathway) or may be triggered indirectly, via a Bcl-2 inhibitable mitochondrial permeabilization event (endogenous pathway). Thymocyte apoptosis is generally accompanied by proteasome activation. If death is induced by DNA damage, inactivation of p53, overexpression of a Bcl-2 transgene, inhibition of protein synthesis, and antioxidants (N-acetylcyteine, catalase) prevent proteasome activation. Glucocorticoid-induced proteasome activation follows a similar pattern of inhibition except for p53. Caspase inhibition fails to affect proteasome activation induced by topoisomerase inhibition or glucocorticoid receptor ligation. In contrast, caspase activation (but not p53 knockout or Bcl-2 overexpression) does interfere with proteasome activation induced by CD95. Specific inhibition of proteasomes with lactacystin or MG123 blocks caspase activation at a pre-mitochondrial level if thymocyte apoptosis is induced by DNA damage or glucocorticoids. In strict contrast, proteasome inhibition has no inhibitory effect on the mitochondrial and nuclear phases of apoptosis induced via CD95. Thus, proteasome activation is a critical event of thymocyte apoptosis stimulated via the endogenous pathway yet dispensable for CD95-triggered death. Cell Death and Differentiation (2000) 7, 368–373
Similar content being viewed by others

Introduction
Apoptosis is almost universally accompanied by the activation of a specific set of cysteine proteases, the caspases,1 as well as by signs of mitochondrial membrane permeabilization including a loss of the inner transmembrane potential (ΔΨm).2,3 The functional hierarchy among these events depends on the apoptosis inducing stimulus. In the ‘exogenous pathway’, caspase activation is triggered as an upstream event, e.g. by recruiting caspases to a complex formed at the plasma membrane, once a so-called death receptor from the CD95/Fas/Apo-1 family has been crosslinked.4 In some cell types (‘type 1 cells’) including thymocytes stimulated via CD95, apical caspases succeed in amplifying the lethal signal via the activation of downstream caspase, and mitochondrial membrane permeabilization (which is triggered by caspases or caspase cleavage products) is dispensable for the death process.5,6 In contrast, most ‘endogenous’ apoptosis triggers (e.g. DNA damage, glucocorticoids) activate caspases in an indirect fashion, namely by first permeabilizing mitochondrial membranes in a Bcl-2-inhibitable fashion.2,3,7 This leads to the translocation of cytochrome c from the mitochondrial intermembrane space to the cytosol where cytochrome c interacts with a molecular complex, the apoptosome, that sets of the caspase activation cascade.3 This scenario explains why Bcl-2, which maintains the barrier function of mitochondrial members, can inhibit apoptosis induced via the endogenous pathway yet fails to protect type 1 cells subjected to CD95 ligation.5,8,9 Moreover, it explains why a series of signal transduction pathways involving p53, protein neosynthesis, or changes in the redox potential only are critical for the endogenous pathway yet totally irrelevant to CD95 induced death.5,7,10,11,12,13
The proteasome ubiquitin pathway is involved in the degradation of short lived regulatory proteins including key effectors of cell cycle and apoptosis.14 Depending on the cellular context and the balance of pro- and anti-apoptotic regulators, inhibition of proteasomes may induce apoptosis (as this is the case in numerous proliferating cell types)15 or prevent apoptosis (as this is the case in cell cycle arrested neurons, thymocytes, or insect muscle cells).16,17,18,19 In thymocytes, proteasome activation is known to be triggered by a wide range of different apoptosis-inducing protocols triggering the endogenous pathway,17 and inhibition of proteasomes can prevent apoptosis at a pre-mitochondrial stage, that is before mitochondria display signs of membrane permeabilization and before caspases are activated.20 Stimulated by these observations, we decided to investigate the relationship between proteasome and caspase activation in the endogenous versus exogenous pathway. The data contained in this paper establish the molecular order between proteasomes and several key apoptosis regulators including p53, Bcl-2, redox imbalances, and caspases. Moreover, they establish that proteasome activation generally accompanies thymocyte apoptosis. However, proteasomes are only required for cell death induced via the endogenous pathway yet are dispensable for CD95-triggered apoptosis.
Results and Discussion
Proteasome activation induced by DEX or etoposide requires RNA/protein synthesis and ROS signaling but does not rely on caspase activation
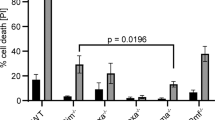
Thymocyte apoptosis was triggered by the glucocorticoid receptor agonist DEX and the topoisomerase inhibitor etoposide. These agents caused an increase in proteasome activity, as determined by means of the fluorescent substrate N-succinyl-L-leucyl-L-leucyl-L-valyl-L-tyrosine-7-amido-4-methylcoumarin21 (Figure 1). The RNA synthesis inhibitor actinomycin D, the protein synthesis inhibitor cycloheximied (CHX), antioxidants such as N-acetylcysteine (NAC) or catalase, or the glucocorticoid receptor antagonist RU38486 (which does not interfere with etoposide-induced cell death) prevent both the signs of nuclear apoptosis (Figure 1B) and proteasome activation (Figure 1C). As a result, upstream signals (macromolecule synthesis, antioxidant sensitive events) triggered by glucocorticoid receptor occupancy or topoisomerase II inhibition are required for both proteasome activation and induction of nuclear DNA loss. Caspase activation is not required for cell death induced by DEX or etoposide in thymocytes,22 although it is indispensable for nuclear DNA degradation to occur, presumably by interfering with the activation of caspase-dependent DNAse (CAD).23 Inhibition of caspases using the broad spectrum inhibitor Z-VAD.fmk (which results in a complete inhibition of nuclear DNA loss; black columns in Figure 2) fails to prevent proteasome activation (gray columns in Figure 2) induced by DEX or etoposide. As a control, Z-VAD.fmk did prevent both caspase and proteasome activation if CD95/Fas/Apo-1 was employed as an apoptosis inducer (Figure 2), thus underlining the importance of apical caspases interacting with the CD95 receptor complex in CD95-mediated signaling.4,5 Altogether these data place the activation of proteasomes upstream of that of caspases, at least in DEX- or etoposide-induced thymocyte apoptosis.

Stimulation of proteasome activity by etoposide or DEX and its modulation by inhibitors of protein/RNA synthesis or anti-oxidants. Thymocytes were cultured for 4 h in the presence of the indicated agents (gray areas), and proteasome activity was measured in cytosolic extracts using the substrate N-succinyl-L-leucyl-L-leucyl-L-valyl-tyrosine-7-amido-4-methylcoumarin (A). In addition, aliquots of cells were fixed and permeabilized with ethanol and stained with PI to determine the frequency of hypoploid nuclei (B). This experiment was repeated twice yielding similar results. Mean values±S.D. of triplicates are shown

Effect of the caspase inhibitor Z-VAD.fmk on proteasome activation and nuclear apoptosis. Thymocytes were cultured for 4 h in the presence or absence of DEX, etoposide, αCD95 and/or Z-VAD.fmk, followed by determination of proteasome activity and the frequency of hypoploid nuclei. Z-VAD.fmk was added 15 min before the apoptosis inducers
p53 and Bcl-2 function upstream of proteasome activation
In thymocytes, p53 is required for apoptosis induced by DNA damaging agents such as etoposide or γ-irradiation, yet is dispensable for DEX- and CD95-induced cell death.10,11 As shown in Figure 3A, cells from p53 knock-out mice (p53−/−) did not only fail to undergo nuclear apoptosis but also lacked proteasome activation in response to DNA damage. In contrast, no significant difference in proteasome activation was detected between p53−/− and control thymocytes (p53+/+) treated with DEX or αCD95. Overexpression of Bcl-2 in thymocytes causes a different pattern of apoptosis inhibition. It inhibits cell death induction by DEX and etoposide, yet has no influence on CD95-triggered apoptosis (Figure 3A). Again, a close correlation between nuclear apoptosis and proteasome activity was detected. Overexpression of Bcl-2 prevents activation of the proteasome induced by DEX or etoposide and only has a moderate inhibitory effect on that triggered via CD95. These data place p53 and Bcl-2 upstream of proteasomes in those pathways in which they act as indispensable pro-apoptotic signals (the case of p53 in DNA damage-induced cell death) or as anti-apoptotic compounds (the case of Bcl-2 in DEX or DNA-damage-induced apoptosis), respectively.

Effect of p53 and Bcl-2 on proteasome activation. Thymocytes from p53−/− mice (A) or mice carrying a Bcl-2 transgene (B), as well as thymocytes from suitable wild type controls, were subjected to the indicated apoptosis inducing treatment. After 4 h of culture, the activity of proteasomes and the loss of nuclear DNA was quantified. Results are representative of three independent determinations
CD95-induced thymocyte apoptosis does not require proteasome activation
DEX- or etoposide-induced apoptosis can be blocked by the specific proteasome inhibitors MG132 and lactacystin,17 as well as LLnL, an inhibitor of proteasomes and calpain17,21 (20 and Figure 4). Surprisingly, however, none of these inhibitors prevented the nuclear apoptosis induced by CD95 crosslinking (Figure 4B), although, as an internal control, they did prevent the increase of proteasome activity induced by αCD95 (Figure 4A). Similarly, proteasome inhibition effectively blocked the activation of a caspase-3-like enzyme cleaving the caspase substrate DEVD.afc, if this activity was induced by DEX or etoposide (Figure 4B). However, no suppressive effect of MG132, lactacystin, or LLnL was detected if caspase-3-like activation was stimulated via CD95 (Figure 4B). We have previously shown that proteasome activation was required for the ΔΨm dissipation and the mitochondrial generation of superoxide anion induced by DEX or etoposide.20 As shown in Figure 4D, proteasome inhibition however failed to affect these mitochondrial hallmarks of apoptosis, if induced by CD95. In conclusion, proteasome activation is not required for CD95-mediated caspase activation, nuclear DNA loss, ΔΨm dissipation, or mitochondrial superoxide anion generation.

Effects of three proteasome inhibitors (MG132, lactacystin, and LLnL) on thymocyte apoptosis induced by DEX, etoposide, or αCD95. Cells were cultured for 4 h in the presence of the indicated combination of agents, followed by quantitation of proteasome activity (A), nuclear apoptosis (B), or cleavage of the caspase-3 substrate Ac-DEVD.afc (C). In addition, cells were stained with DiOC6(3) and HE and analyzed by cytofluorometry (D), as described in Materials and Methods. Numbers refer to the percentage of cells found in each quadrant
Concluding remarks
The data contained in this paper, as well as those from previous reports,17,20 establish the position of proteasomes with respect to a number of different pro-apoptotic molecules activated in the endogenous and exogenous (CD95-triggered) pathways leading to thymocyte death (Figure 5). The synthesis of new proteins (which is blocked by CHX and indirectly by actinomycin D), as well as oxidative reactions (blocked by the glutathione precursor N-acetylcysteine or catalase), which are involved in the endogenous pathway triggered by DEX or DNA damage, act upstream of the proteasome (Figure 1), the activation of which, in turn, is required for mitochondrial membrane permeabilization,20 caspase activation (Figure 4C), and nuclear apoptosis (Figure 4B). Similarly, the DNA damage sensor p53 acts upstream of the proteasome and is required for its activation by etoposide or γ-irradiation (but not by DEX) (Figure 3A). In contrast, caspase inhibition does not prevent proteasome activation induced by DNA damage or DEX (Figure 2). We have shown in the past that bongkrekic acid, a ligand of mitochondrial protein that stabilizes the ΔΨm, does not affect activation of the proteasome yet prevents downstream caspase activation and nuclear apoptosis.20 This observation, together with the fact that proteasome inhibition could inhibit the ΔΨm loss, led us to postulate that proteasome activation occurred upstream of mitochondria.20 Unexpectedly, however, Bcl-2 (which is thought to primarily act on mitochondria24,25) not only prevents the ΔΨm dissipation but also suppresses the activation of proteasomes triggered via the endogenous pathway (Figure 3B). This hints to a close connection between mitochondrial membrane permeabilization and proteasome activation (as suggested in Figure 5), or alternatively suggests that Bcl-2 (which also localizes to extramitochondrial membranes) may have pleiotropic effects that directly or indirectly affect proteasome function.

Molecular ordering of apoptotic events in thymocyte apoptosis. Based on the effect of different inhibitors or genetic manipulations at least two major pathways may be delineated. For details and references consult main text
More intriguingly, it appears that primary activation of caspases by CD95 crosslinking causes apoptosis via a pathway that is completely resistant to proteasome inhibitors (Figure 4). Thus, although the proteasome is activated after CD95 ligation, presumably in a caspase dependent fashion (as suggested by the inhibitory effect of Z-VAD.fmk; Figure 2), this activation has no impact whatsoever on the nuclear (Figure 4B) and mitochondrial (Figure 4D) signs of apoptosis induced by CD95. Altogether these results indicate that proteasome activation is an important mechanism of thymocyte apoptosis induction via the endogenous pathway yet completely dispensable for death activation via the prototypic exogenous CD95 pathway.
Materials and Methods
Animals and cell culture conditions
Thymocytes from female 4–6 week-old Balb/c mice were cultured in RPMI 1640 medium supplemented with 10% fetal calf serum, L-glutamine, and antibiotics. In addition, thymocytes were obtained from C57Bl/6 mice rendered deficient for p53 by homologous recombination (p53−/−)26 or mice overexpressing Bcl-2 under the control of the immunoglobulin heavy chain enhancer27 were obtained from the central animal facility of CNRS (Orléans, France).
Induction and inhibition of apoptosis
Cells (1–5×106 ml) were cultured in a humidified atmosphere containing 5% CO2 at 37°C during the indicated interval, alone or with combinations of the following agents: dexamethasone (DEX; 1 μM; Sigma, St. Louis, MO, USA), etoposide (VP-16, 10 μM, Sigma), irradiation (10 Gy; Gravaton industries, RX30/55 irradiator, Gosport, UK), an antibody specific for CD95/Apo-1/Fas (clone 154000D, 500 ng/ml; PharMingen, San Diego, CA, USA), MG132 (CBZ-leucyl-leucyl-leucinal; 30 μM; Peptides International, Louisville, KY, USA), lactacystin (30 μM; purchased from Dr. MR Corey, Harvard University, Cambridge, MA, USA), N-acetyl-L-leucinyl-L-leucinal-L-norleucinal (LLnL, 30 μM; also called ‘calpain inhibitor I’; Bachem, Basel, Switzerland), N-benzyloxycarbonyl-Val-Ala-Asp-fluoromethylketone (Z-VAD.fmk; 50 μM, Enzyme Systems, Dublin, CA, USA), N-acetylcysteine (30 mM), catalase (1000 U/ml), actinomycin D (1 μM), cycloheximide (CHX, 35 μM; Sigma), and/or RU38486 (10 μM, kindly provided by Roussel Uclaf, Romainville, France).
Determination of apoptosis-associated parameters
In accord with published protocols,28 two fluorochromes were employed to determine apoptosis-associated changes: 3,3′dihexyloxacarbocyanine iodide (DiOC6(3), 20 nM, 15 min, 37°C) for ΔΨm quantification and hydroethidine (HE, 4 μM, 15 min, 37°C) for the determination of superoxide anion generation causing oxidation of He to ethidium (Eth). Cytofluorometry was performed on a Coulter Elite II analyzer. Fluorescence was registered for all cells (large and apoptotic), while excluding debris. The frequency of subdiploid cells was determined by propidium iodide staining of ethanol-permeabilized cells.29 The capacity of cytosols to cleave the caspase-3 recognition site DEVD was measured using Ac-DEVD-amino-4-methylcoumarin (Bachem, Basel, Switzerland) as fluorogenic substrate, as described.7
Determination of proteasome activity
Proteasome activity was determined by means of the cell-permeable fluorogenic substrate N-succinyl-L-leucyl-L-leucyl-L-valyl-L-tyrosine-7-amido-4-methylcoumarin (Bachem, Basel, Switzerland), following published protocols.21 Cells (4×106 in 200 μl, PBS, pH=7.4) were incubated during 30 min at 37°C with this substrate (100 μM), and the 7-amino-4-methylcoumarin fluorescence generated by its cleavage was quantitated in a Kontron SFM 25 spectrofluorometer (Kontron AG, Zurich, Switzerland). Background values of non-stimulated cells were not reduced by the proteasome inhibitors MG132 (30 μM) or lactacystin (30 μM) and thus were subtracted from the experimental values.
Abbreviations
- CHX:
-
cycloheximide
- ΔΨm:
-
mitochondrial transmembrane potential
- DEX:
-
dexamethasone
- Eth:
-
ethidium
- DiOC6(3):
-
3,3′dihexyloxacarbocyanine iodide
- HE:
-
hydroethidine
- LLnL:
-
N-acetyl-L-leucinyl-L-leucinal-L-norleucinal (=calpain inhibitor I)
- NAC:
-
N-acetylcysteine
- PI:
-
propidium iodide
- PT:
-
permeability transition
- Z-VAD.fmk:
-
N-benzyloxycarbonyl-Val-Ala-Asp-fluoromethylketone
References
Nicholson DW and Thornberry NA . (1997) Caspases: killer proteases. Trends Biochem. Sci. 22: 299–306
Kroemer G, Dallaporta B and Resche-Rigon M . (1998) The mitochondrial death/life regulator in apoptosis and necrosis. Annu. Rev. Physiol. 60: 619–642
Green DR and Reed JC . (1998) Mitochondria and apoptosis. Science 281: 1309–1312
Medema JP, Scaffidi C, Kirschkel FC, Shevchenko A, Mann M, Krammer PH and Peter ME . (1997) FLICE is activated by association with the CD95 death-inducing signaling complex (DISC). EMBO J. 16: 2794–2804
Scaffidi C, Fulda S, Srinivasan A, Friesen C, Li F, Tomaselli KJ, Debatin K-M, Krammer PH and Peter ME . (1998) Two CD95 (APO-1/Fas) signaling pathways. EMBO J. 17: 1675–1687
Yin X-M, Wang K, Gross A, Zhao Y, Zinkel S, Klocke B, Rothe KA and Korsmeyer SJ . (1999) Bid-deficient mice are resistant to Fas-induced hepatocellular apoptosis. Nature 400: 886–891
Susin SA, Zamzami N, Castedo M, Daugas E, Wang H-G, Geley S, Fassy F, Reed J and Kroemer G . (1997) The central executioner of apoptosis. Multiple links between protease activation and mitochondria in Fas/Apo-1/CD95- and ceramide-induced apoptosis. J. Exp. Med. 186: 25–37
Strasser A, Harris AW, Huang DCS, Krammer PH and Cory S . (1995) Bcl-2 and Fas/APO-1 regulate distinct pathways to lymphocyte apoptosis. EMBO J. 14: 6136–6147
Huang DCS, Cory S and Strasser A . (1997) Bcl-2, Bcl-XL, and adenovirus protein E1B19kD are functionally equivalent in their ability to inhibit cell death. Oncogene 14: 405–414
Clarke AR, Purdie CA, Harrison DJ, Morris RG, Bird CC, Hooper ML and Wyllie AH . (1993) Thymocyte apoptosis induced by p53-dependent and independent pathways. Nature 362: 849–852
Lowe SW, Schmitt EM, Smith SW, Osborne BA and Jack T . (1993) p53 is required for radiation-induced apoptosis in mouse thymocytes. Nature 362: 847–849
Castedo M, Hirsch T, Susin SA, Zamzami N, Marchetti P, Macho A and Kroemer G . (1996) Sequential acquisition of mitochondrial and plasma membrane alterations during early lymphocyte apoptosis. J. Immunol. 157: 512–521
Macho A, Hirsch T, Marzo I, Marchetti P, Dallaporta B, Susin SA, Zamzami N and Kroemer G . (1997) Glutathione depletion is an early and calcium elevation a late event of thymocyte apoptosis. J. Immunol. 158: 4612–4619
Orlowski RZ . (1999) The role of the ubiquitin-proteasome pathway in apoptosis. Cell Death Differ. 6: 303–313
Adams J, Palombella VJ, Sausville EA, Johnson J, Destree A, Lazarus DD, Maas J, Pien CS, Prakash S and Elliott PJ . (1999) Proteasome inhibitors: A novel class of potent and effective antitumor agents. Cancer Res. 59: 2615–2622
Jones MEE, Haire MF, Kloetzel PM, Mykles DL and Schwartz LM . (1995) Changes in the structure and function of the multicatalytic proteinase (proteasome) during programmed cell death in the intersegmental muscles of the hawkmoth, Manduca sexta. Dev. Biol. 169: 436–447
Grimm LM, Goldberg AL, Poirier GG, Schwartz LM and Osborne BA . (1996) Proteasomes play an essential role in thymocyte apoptosis. EMBO J. 15: 3835–3844
Sadoul R, Fernandez PA, Quiquerez AL, Martinou I, Maki M, Schroter M, Becherer JD, Irmler M, Tschopp J and Martinou JC . (1996) Involvement of the proteasome in the programmed cell death of NGF-deprived sympathetic neurons. EMBO J. 15: 3845–3852
Hastings RA, Eyheralde I, Dawson SP, Walker G, Reynolds SE, Billett MA and Mayer RJ . (1999) A 220-kDa activator complex of the 26 S proteasome in insects and humans. A role in type II programmed insect muscle cell death and cross-activation of proteasomes from different species. J. Biol. Chem. 274: 25691–25700
Hirsch T, Dallaporta B, Zamzami N, Susin SA, Ravagnan L, Marzo I, Brenner C and Kroemer G . (1998) Proteasome activation occurs at an early, pre-mitochondrial step of thymocyte apoptosis. J. Immunol. 161: 35–40
Reidlinger J, Pike AM, Savory PJ, Murray RZ and Rivett AJ . (1997) Catalytic properties of 26 S and 20 S proteasomes and radiolabeling of MB1, LMP7, and C7 subunits associated with trypsin-like and chymotrypsin-like activities. J. Biol. Chem. 272: 24899–24905
Hirsch T, Marchetti P, Susin SA, Dallaporta B, Zamzami N, Marzo I, Geuskens M and Kroemer G . (1997) The apoptosis-necrosis paradox. Apoptogenic proteases activated after mitochondrial permeability transition determine the mode of cell death. Oncogene 15: 1573–1582
Enari M, Sakahira H, Yokoyoma H, Okawa K, Iwamtsu A and Nagata S . (1998) A caspase-activated DNase that degrades DNA during apoptosis, and its inhibitor ICAD. Nature 391: 43–50
Kroemer G . (1997) The proto-oncogene Bcl-2 and its role in regulating apoptosis. Nature Med 3: 614–620
Gross A, McDonnell JM and Korsmeyer SJ . (1999) Bcl-2 family members and the mitochondria in apoptosis. Genes Dev 13: 1899–1911
Donehower LA, Harvey M, Slagle BL, McArthur MJ, Montgomery CA, Butel JS and Bradley A . (1992) Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumors. Nature 356: 215–221
Strasser A, Harris AW and Cory S . (1991) bcl-2 transgene inhibits T cell death and perturbs thymic self-censorship. Cell 67: 889–899
Zamzami N, Marchetti P, Castedo M, Decaudin D, Macho A, Hirsch T, Susin SA, Petit PX, Mignotte B and Kroemer G . (1995) Sequential reduction of mitochondrial transmembrane potential and generation of reactive oxygen species in early programmed cell death. J. Exp. Med. 182: 367–377
Nicoletti I, Migliorati G, Pagliacci MC and Riccardi C . (1991) A rapid simple method for measuring thymocyte apoptosis by propidium iodide staining and flow cytometry. J. Immunol. Meth. 139: 271–280
Acknowledgements
Supported by a special grant by the Ligue National contre le Cancer, as well as by grants from ANRS, FRM (to G Kroemer), Assistance Publique-Hôpitaux de Paris and CANAM (contract 98006 to E Daugas). M Loeffler receives a fellowship from the Austrian Science Foundation.
Author information
Authors and Affiliations
Corresponding author
Additional information
Edited by L Fesus
Rights and permissions
About this article
Cite this article
Dallaporta, B., de Pablo, M., Maisse, C. et al. Proteasome activation as a critical event of thymocyte apoptosis. Cell Death Differ 7, 368–373 (2000). https://doi.org/10.1038/sj.cdd.4400661
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.cdd.4400661
Keywords
This article is cited by
-
BAC-mediated transgenic expression of fluorescent autophagic protein Beclin 1 reveals a role for Beclin 1 in lymphocyte development
Cell Death & Differentiation (2008)
-
Proteasome inhibition drastically but reversibly impairs murine lymphocyte development
Cell Death & Differentiation (2008)
-
Regulation of differential pro- and anti-apoptotic signaling by glucocorticoids
Apoptosis (2007)
-
Activation of NFκB and Ub-proteasome pathway during apoptosis induced by a serum factor is mediated through the upregulation of the 26S proteasome subunits
Apoptosis (2006)
-
Apoptosis by gemcitabine in non-small cell lung cancer cell line KNS62 is induced downstream of caspase 8 and is profoundly blocked by Bcl-xL over-expression
Langenbeck's Archives of Surgery (2005)
