
Abstract
Two preneoplastic cell lines have been utilized to study changes in the regulation of apoptosis during neoplastic progression [sup+I (stage I) and sup−II (stage II)]. Sup+I cells are prone to undergo apoptosis, while sup−II cells are relatively resistant. We report that induction of apoptosis in sup+I cells is tightly correlated with the formation of c-Fos/p300 complexes, which were not present in the non-apoptotic sup−II cells under the same conditions. When apoptosis was induced in the sup−II cells by over-expression of c-Fos, concomitant c-Fos:p300 complexes were detected. Over-expression of p300 resulted in apoptosis in sup−II cells and also in p53wt human tumor cells, but not in p53mutant human tumor cells. Over-expression of the C-terminal fragment of p300, which contains the c-Fos binding site, enhanced apoptosis, suggesting that the c-Fos:p300 complex is actively involved in apoptosis. We propose that p300 could function as a general mediator of transcription factor-induced apoptosis.
Similar content being viewed by others


Introduction
Central to our understanding of biochemical changes associated with neoplastic progression is the identification of gene products that have the potential to contribute to the deregulation of the cell cycle and of apoptosis. A tumor can develop if proto-oncogenes involved in cell cycle progression become inappropriately expressed at high levels, resulting in uncontrolled cellular growth. However in some cases, cell death pathways may become altered and a tumor can develop, even though cell growth controls remain normal. Our studies are directed toward understanding the changes in the regulation of apoptosis during neoplastic progression.1 We found that increased levels of c-Fos protein, a component of the activating protein-1 (AP-1) transcription factor, was responsible, at least in part, for sensitizing early preneoplastic cells to undergo apoptotic cell death.2 Further, we found that later stage neoplastic cells that had evolved to bypass apoptotic signals could be induced to undergo apoptosis by over-expression of c-Fos. c-Fos is associated with apoptosis in a number of different systems.3,4,5,6,7,8,9,10,11,12 In fact, a number of transcription factors are involved in modulating apoptosis, such as E2F1, NF-κB, B-Myb, c-Myb, EGR-1, Stat3, Tax, and c-Jun.13,14,15,16,17,18,19,20,21,22,23,24,25,26 NF-κB and c-Myb have been shown to prevent apoptosis in certain cell types, while E2F1, EGR-1, Stat3, Tax, and c-Jun are implicated in the activation of apoptosis. Interestingly, activation of transcription may or may not be involved, depending on the transcription factor and the cell type.27,28,29,30,31,32 For example, we have shown that c-Fos mediates apoptosis without activation of transcription.2 In this case, c-Fos, and maybe transcription factors in general, may function as a repressor of transcription, and specific protein partners could dictate this function. Interestingly, of the transcription factors mentioned above, most physically interact with p300/CBP.18,33,34,35,36,37,38,39,40,41
p300 was originally identified by its ability to associate with the adenovirus E1A oncoprotein.42,43 P300 and the closely related CREB binding protein (CBP) act as transcriptional co-activators.44,45,46,47 CBP and p300 interact with common specific proteins, and it was originally thought that they were functionally similar. However, potentially important differences are being unraveled.48 Knockout mouse studies indicate that p300 and CBP, at least in part, are functionally unique during development.49 Some studies have suggested an association between p300 and apoptosis in virally transformed cells. E1A-induced immortalization results in increased susceptibility to induction of apoptosis,50,51 and both the transforming ability and apoptosis inducing ability of E1A require the capability to bind p300/CBP.52,53 Apoptosis occurs when virally transformed cells are placed in anti-proliferative conditions,53 and it is suggested that viral repression of host genes, to facilitate viral growth, results in apoptosis as an undesirable side effect.51 There is a growing body of evidence that genetic alterations in p300 and/or CBP genes might be linked to the onset of human tumors,54,55 implying that p300/CBP acts as a tumor suppressor. Remarkably, the mutations were located within the second and third Cys/His-rich regions of p300, one of which is the c-Fos binding region.
The studies presented here investigate the mechanism of c-Fos-induced apoptosis. Our earlier data indicated that c-Fos does not function as a transcriptional activator in the apoptotic process.2 In the presence of protein synthesis inhibitors, apoptosis could still be induced by activation of a FosER chimeric protein, (c-Fos/estrogen ligand binding domain fusion protein). p55v-fos, which is highly proficient in transcriptional activation but deficient in the transcriptional-repression activity, could not substitute functionally to induce apoptosis in our system, which again indicated that transcriptional activation was not the required function for induction of apoptosis. Inasmuch as p300 associates with c-Fos,34 we hypothesized that the mechanism of c-Fos-induced apoptosis involves an interaction with p300.
For these studies we utilized two preneoplastic cell lines from a series of Syrian hamster embryo cell lines that have been extensively characterized and that represent different stages in the neoplastic process. The first cell line, isolated as an immortal, non-tumorigenic clone, is representative of early stage, preneoplastic cells and has been characterized as ‘tumor-suppressor gene plus’ (sup+I), since these cells retain the ability to suppress tumorigenicity when hybridized with tumor cells. The second cell line, termed ‘tumor suppressor gene minus’ (sup−II), is representative of a later stage of preneoplastic cells. Although sup−II cells are still non-tumorigenic, they no longer have the capability to suppress tumorigenicity in cell hybrids.56 The tumor suppressor gene altered in these cells has not been conclusively identified, although the cells differ in their expression of putative tumor suppressor genes, H19 and tropomyosin 1.57 The RB and p53 genes are wild-type in both cell types.58 As previously reported, contrasting responses were observed when these two cell lines were placed under growth factor deprivation conditions; sup+I cells were highly susceptible to apoptosis, whereas, sup−II cells were resistant.1
To investigate the role of p300 in the events leading to c-Fos-induced apoptosis sup+I, sup−II, sup−II /FosER, sup−II /v-Fos cells were placed in low serum conditions. At multiple time-points, immune complexes containing c-Fos and associated proteins were analyzed for the presence of p300. We found that a transient complex was formed between c-Fos and p300 in cells that died by apoptosis, sup+I and sup−II /FosER, but not in cells that are resistant to death, sup−II and sup−II /v-Fos. In addition, we report that over expression of p300 resulted in activation of apoptosis, through a p53-dependent mechanism. Because p300 interacts with multiple transcription factors that are activated through diverse signal transduction pathways, our findings lead us to suggest that p300 is a mediator of signals to activate apoptosis. We propose that p300 acts as a ‘sensor’, which can detect mixed cellular signals, i.e., signals to proliferate coincident with signals to growth arrest.
Results
Endogenous c-Fos / p300 immune complexes are present in cells undergoing low-serum-induced apoptosis
The high propensity to undergo apoptotic death in the early preneoplastic cells (sup+I) has been linked with increased expression of endogenous c-Fos, implicating a role for c-Fos.2 We investigated whether c-Fos and p300 could be detected in a specific protein complex in sup+I cells induced to undergo apoptosis or sup−II cells under the same conditions. Cell extracts of sup+I and sup−II cells in 0.1% serum were isolated at 0, 20, 40, 60, 80, and 100 min. Protein complexes were immunoprecipitated with either p300 or c-Fos antibody. Western blot analyses of the anti-p300 precipitates from the sup+I cells revealed the presence of a discrete fraction of c-Fos transiently associated with p300 at the 100 min time-point (Figure 1A). Further, p300 could be detected in anti-c-Fos immunoprecipitated complexes (Figure 1B). The transient complex was not observed in sup−II cells, which do not undergo apoptosis under these conditions (Figure 1C). In a repeat experiment, the time-points were extended to 2 h in order to determine if the complex was transient or if it persisted over time. In this experiment the c-Fos:p300 complex was detected maximally at 90 min, diminished by 100 min, and was not present at later time-points up to 4 h. The complex was not detected in the sup−II cells (Figure 1E). DNA fragmentation could be observed in sup+I cells as early as 4 h in low serum and no DNA fragmentation was observed in the sup−II cells (Figure 2). The timely appearance of DNA fragmentation 2.5 h after the p300-c-Fos complex was observed suggests that the complex is mediating apoptotic signals.

c-Fos and p300 complexes are detected in preneoplastic sup+I in low serum, but not in sup−II cells. (A, C, D and E) p300 was immunoprecipitated and complexes were analyzed for the presence of c-Fos by Western blot. Endogenous c-Fos protein was detected at 100 min in low serum. Blots A and C were reprobed with p300 antibody to show equal amounts of p300 were immunoprecipitated. A nonspecific (NS) mouse monoclonal was used as an antibody control. (B). c-Fos was immunoprecipitated and complexes were analyzed for the presence of p300 by Western blot. The NS antibody control is peptide competed anti-cFos. HeLa cell nuclear lysates were used as a marker for p300

DNA fractionation on a 1% agarose gel. sup+I and sup−II cells were placed in 0.1% serum. DNA laddering was first detected in sup+I cells at 4 h. No DNA fragmentation was observed in sup−II cells. Sup−II /FosER cells were incubated in serum-free medium plus E2 and DNA fragmentation by was detected after 8 h
Induction of apoptosis by over-expression of c-FosER is associated with an interaction with p300
Later-stage preneoplastic cells (sup−II) can be induced to undergo apoptosis in low serum by exogenously forced over-expression of the chimeric c-FosER protein.2 Under normal conditions, the chimeric protein is constitutively expressed and resides in the cell in an inactive conformation until estradiol (E2) is added to the medium. Once E2 binds to the estrogen ligand-binding domain (ER) of the chimeric protein, the conformation of the protein changes, resulting in an active form of c-Fos and, as a consequence, the cells undergo apoptosis. If the c-Fos:p300 complex is part of the apoptosis-inducing cascade, then a c-FosER/p300 complex should be observed in the sup−II /FosER cells, when induced to undergo apoptosis. The first experiments were designed to optimize for detection of the c-FosER/p300 complex by first incubating the cells in phenol red-free, serum free medium for 30 min, then E2 (1 μM) was added and cells were harvested at 10, 15, 20, 30, and 45 min. The complex was observed at the 20 min time-point (data not shown). Once the time requirements were established, the Sup−II/FosER cells and sup−II/v-Fos cells were examined under multiple conditions to establish whether or not the formation of the c-FosER/p300 complex could be tightly associated with cell death (Figure 3A). First the cells were placed in serum free medium for 24 h and then serum stimulated for 1 h to increase the endogenous c-Fos protein and to enhance for a strong proliferative response (Figure 3A, lanes 1 and 7). The complex was not observed in either cell line. Also analyzed were logarithmically growing cells, (lanes 2 and 8), logarithmic cells plus E2 for 20 min (lanes 3 and 9), cells in serum free medium alone for 30+20 min (lanes 4 and 10), and serum free medium for 30 min plus 20 min with E2 (lanes 5 and 11). The only time in which the anti-c-Fos antibody immunoprecipitated complexes contained p300 was in conditions shown to induce apoptosis in the sup−II/FosER cells, i.e., serum free medium plus E2 (Figure 3A, lane 5). The v-Fos expressing cells do not die in low serum (plus E2 or minus E2)2 and the complex was not observed (Figure 3A, lane 11). Figure 3A, lanes 6 and 12 are anti-p300 immunoprecipitations followed by anti-p300 westerns to show that this antibody does bind to p300 and is present in similar amounts in these cells. HeLa cell extract was used as a marker for p300 (lane 13).

(A) c-FosER activation in low serum results in c-FosER/p300 complex. Analysis of c-FosER and v-Fos immunoprecipitated complexes for the pesence of p300 by Western blot: lanes 1 and 7, Sup−II/FosER cells and sup−II/v-Fos cells incubated in serum free medium for 24 h and then serum stimulated for 1 h; lanes 2 and 8, logarithmically growing cells; lanes 3 and 9, logarithmic cells plus E2 for 20 min; lanes 4 and 10, cells in serum free medium alone for 50 min; lanes 5 and 11, serum free medium for 30 min plus 20 min with E2. Lanes 6 and 12 are anti-p300 immunoprecipitations followed by anti-p300 Westerns to show antibody binding to p300. HeLa cell extract was used as a marker for p300 (lane 13). (B) Complex formation between exogenously expressed c-FosER and p300 in sup−II /FosER cells. Immunoprecipitations were performed using p300 monoclonal antibodies (UBI). A NS mouse monoclonal was used as an antibody control. c-Fos specific Western blot analysis showed c-FosER was detectable in p300 immune complexes, 60 min after induction of apoptosis. (C) FosER protein was detected in immune complexes with p300-HA protein that were immunoprecipitated using a HA-tag specific antibody (12CA5)
To mimic the experimental protocols utilized to study endogenous c-Fos:p300 complexes, the sup−II/FosER cells were placed in serum-free medium and the E2 was added immediately. Cells were harvested (10, 20, 30, 40, 50, 60 and 80 min) and were immunoprecipitated with anti-p300 antibody. Western blot analysis of the immune complexes revealed the presence of the 80 kD FosER fusion protein associated with p300 at 60 min (Figure 3B). A nonspecific (NS) mouse monoclonal antibody (anti-HSP70) was utilized as a control. To confirm the in vivo interaction between p300 and c-FosER, sup−II/FosER cells were transiently transfected with a HA-tagged p300 vector. At 18 h post-electroporation, cells were placed in serum-free medium for 30 min. c-FosER was activated by addition of E2 to the culture medium for 20 min. Cell extracts were immunoprecipitated using an anti-HA antibody (12CA5 Mab) and precipitated complexes were analyzed by Western blot probed for c-FosER. C-FosER co-precipitated with the HA-tag-specific antibody (Figure 3C), thus demonstrating the specificity of the c-Fos:p300 interaction. The time between the observed c-FosER-p300 complex and DNA laddering (Figure 2B) was 6–7 h, longer than the time observed in the sup+I cells of 1–2 h.
Nonetheless, the fact that the phenotype of these cells can be reversed by increasing the levels of c-FosER and that c-FosER-p300 complexes are observed coincident with apoptosis would imply that the mechanism involved in apoptosis is similar in the two cell lines.
Transient over-expression of p300 induces apoptosis in p53 wild-type cells
To address the question of the functional significance of the transient p300/c-Fos complex, it was hypothesized that if the complex is involved in mediating apoptosis, over-expression of p300 would enhance the frequency of apoptosis. Various cell lines were transiently transfected with either a vector containing the β-galactosidase gene or the CMVp300 vector. Apoptosis was measured after 24 h by FACScan analysis of TUNEL labeled cells. Transfection efficiency was estimated as the number of β-gal positive cells in controls. Over-expression of p300 resulted in the induction of apoptosis in cells incubated in 10% serum within 24 h after transfection (Table 1), with an incidence of ∼23% apoptosis in sup+I cells and ∼14% in sup−II cells. There are reports of successful transient transfection experiments in studies of p300,59 although some have reported that stable clones over-expressing p300 could not be established.60 One explanation could be that cells expressing wild-type p53 do not tolerate p300 over-expression. We found that p300 over-expression induced apoptosis in both sup+I and sup−II cells and they both express wild-type p53 protein.58 In addition, we have shown that forced apoptosis through over-expression of c-Fos is p53 dependent.2 This is consistent with the recent reports indicating that p300 can modulate p53 functions.61,62 To test this further, two HT1080 cell lines (human fibrosarcoma) were utilized: one characterized as expressing a normal p53 protein, and the other shown to be a variant that expresses a mutant form of p53 protein. Determination of p53 status was determined by antibody binding specificities and DNA sequencing (Voyta et al., manuscript in preparation).
FACScan analysis of TUNEL labeled cells is plotted as propidium iodide fluorescence on the Y-axis (FL2-A), indicative of DNA content, and FITC fluorescence on the X-axis (FL-1), indicative of labeled apoptotic cells. The terminal transferase enzyme (TdT) was omitted (using one-half of the sample) in order to estimate background fluorescence (Figure 4A,C,E and G). Gates were set so that <3% of the cells were in the FITC positive range. The remainder of the cell sample was labeled to detect apoptotic cells by addition of TdT to the reaction mixture and (Figure 4B,D,F and H). HT1080/p53wild-type cells transfected with β-gal gene show 12.3% apoptotic cells, while those cells transfected with p300 show a frequency of 55.4%. However, HT1080/p53mutant cells were not induced to undergo apoptosis. Interestingly, Western blot analysis of these cells (Figure 5) shows lower levels of endogenous p300 in the HT1080/p53wild-type cells with no detectable increase in p300 protein 24 h post transfection in, implying that those cells expressing the exogenous p300 were lost due to apoptosis. Endogenous p300 levels were higher in HT1080/p53mutant cells, indicating that mutations in p53 permit cellular tolerance of higher levels of p300, and in fact, slightly higher levels are detected in the p300 transfected HT1080/p53mutant cells (Figure 5). In support of this hypothesis, it was determined if the p53wild-type human colorectal carcinoma cell line (RKO) was unable to tolerate over-expression of p300 (Table 1). RKO cells transfected with p300 showed 35% apoptotic cells as compared to 3.7% in the pSV2neo transfected control cells. These data implicate p300 as an important component of the p53-dependent apoptotic pathway.

Exogenously expressed p300 results in activation of apoptosis in p53wt human fibrosarcoma cells (HT1080). FACSan analysis of TUNEL labeled cells: (A, C, E, and G) show data using half of each cell sample containing all components of a TUNEL-labeling reaction except for the terminal transferase (TdT), to set the vertical limits for background fluorescence. (B,D,F, and H) (+TdT) per cent of apoptotic cells. (D) An increase in apoptosis to 55.4% was observed in p53wt cells. (H) No increase was observed in the p53mutant cells

Human tumor cells (HT1080) with a mutant p53 have higher levels of endogenous p300 and appear to tolerate higher levels of exogenously expressed p300. Western blot analysis (1×105 cells/lane) of p300 protein 24 h post-transfection, using monoclonal p300 antibody (UBI)
Evaluation of the mechanism of c-Fos:p300 associated apoptosis
We hypothesized that if c-Fos is inducing apoptosis through sequestration of p300, then over-expression of the C-terminal fragment of p300 should titrate out the c-Fos molecules and block apoptosis. The third Cys/His-rich region of p300/CBP (amino acids 1572–1818) is the segment that interacts with c-Fos.34 In these experiments we utilized a mutant bearing internal deletion that removed a portion of the Fos-binding, CH3 region of p300, termed 5.3 ATG del33, in addition to fragments of p300 fused to a GST moiety: N-terminal (aa 1–596), middle (aa 744–1571), and C-terminal (aa 1572–2370).46,61 The sup−II /FosER cells were transfected by electroporation with vectors coding for β-galactocidase, p300, p300del33, and three p300-fragment fusion proteins. Cells were replated in 10% serum for 8 h. A portion of the cells was placed in serum-free medium plus E2 for an additional 16 h and the remainder of cells was left in 10% serum plus E2. Induction of apoptosis by activation of c-FosER in Sup−II/FosER cells requires the additional stress of serum depletion. The 10% serum plus E2 group was utilized as a control in these experiments, with the rationale that in these cells c-FosER would be active and available to interact p300, but in the absence apoptotic signals, thus permitting us to determine if activation of apoptotic signal transduction pathways is required for induction of the c-Fos:p300 complex. Both floating and attached cells were harvested 24 h after transfection. Apoptosis was monitored by FACS analysis of TUNEL labeled cells (Figure 6). As described above, the cells from each transfection were divided and 1/2 was incubated in all the TUNEL labeling reagents except for TdT (Figure 6A,C). Estimated background fluorescence was then gated and the gates were superimposed onto the analysis of positively labeled apoptotic cells. In 10% serum plus E2 controls groups the per cent apoptosis in cells transfected with the whole p300 gene was slightly elevated, compared to β-gal controls (16–23.7%). However, no increase in apoptosis was observed with the p300del33, or the p300 fragments, compared to β-gal controls (Table 2). Similar to previous experiments, the Sup−II/FosER cells, transfected with control plasmid (β-galactosidase), underwent apoptosis in serum-free medium in the presence of E2 (Figure 6B) with 42.6% TUNEL positive cells. When cells transfected with the whole p300 molecule were placed in serum-free medium plus E2, there was an increase in the number of apoptotic cells to 57.1% (15% increase) (Figure 6D). This is a significant increase, when considering that these cells are dying at a high rate due to c-FosER expression and that the size of the p300 vector may hinder transfection efficiency. Expression of p300del33 in serum-free medium resulted in a reduction in apoptosis from 42.6–21.8% (Table 2). Surprisingly, expression of the N-terminal fragment blocked c-FosER-induced apoptosis to 18% (the -TdT controls were performed but are not shown) (Figure 6E). The frequency of apoptosis in the middle fragment transfected cells was comparable to β-gal alone at 39% (Figure 6F). Also unexpected, expression of the C-terminal fragment enhanced apoptosis to 78% (Figure 6G). The data indicate that the C-terminal portion of the p300 protein is actively involved in the induction of apoptosis.

Per cent apoptosis in sup−II /FosER cells transfected with p300 or fragments of the p300 molecule. Cells were placed in serum-free medium plus E2 to induce apoptosis: background fluorescence, (A and C). TUNEL labels cells expressing B-gal control vector (B) or p300 (D). (E, F and G) show TUNEL labeled cells expressing the p300 fragments
An aliquot of cells (5×105) from the transfections described above was harvested in parallel with cells to be analyzed by TUNEL. We examined the effect of exogenous expression of N-terminal, middle, and C-terminal p300 fragments on endogenous p300. Western blot analysis (Figure 7) shows that over-expression of the N-terminal reduced the levels of p300 protein, while the p300 levels in the C-terminal and middle fragment transfected cells were comparable to levels in the β-gal control. Densometric analysis of band intensities from two separate experiments showed a mean difference of 1.65±8.81 between experiments. This was not statistically different from zero, indicating similar results from each experiment. These data have been included to offer one explanation for the block of c-Fos-induced apoptosis by the N-terminal p300 fragment described above. It appears that a reduction in p300 protein is associated with a loss of apoptotic signals. However, it is possible that the N-terminal fragment expressed in these cells is actively blocking the cell death pathway.

Over-expression of the N-terminal fragment of p300 results in a reduction of endogenous p300. Western blot of sup−II /FosER cells 24 h post-transfection with p300 fragments: β-gal control vector, C-Terminal fragment (C); N-terminal fragment (N); middle fragment (M)
Discussion
The studies presented address the general questions of what mechanisms are involved in transcription factor-mediated apoptosis in the absence of their general role as transcriptional activators. We reported that c-Fos was primarily involved in sensitizing early preneoplastic cells to low serum-induced apoptosis in a p53-dependent pathway.2 The present manuscript is a continuation of these studies into the mechanism of c-Fos-induced apoptosis. We report the novel finding that p300 is involved in mediating c-Fos-induced apoptosis. Further we propose that interactions with p300 protein could be a common pathway for many of the transcription factor-mediated apoptotic pathways. In fact, recent reports describe a role for p300 in the apoptotic activity of another transcription factor, E2F-1.18
In as much as the transient complex was observed only in cells induced to undergo apoptosis, it appears that p300 is actively involved in mediating c-Fos-induced apoptotic cell death. It is important to note that studies presented here include data showing interactions between endogenous c-Fos and endogenous p300, and that this complex was observed after apoptosis was induced but before the cells died. In conjunction with endogenous protein studies, exogenous over-expression of an ER- tagged c-Fos fusion protein resulted in apoptosis and a coincident increase in the level of precipitable c-FosER-p300 complexes. Over-expression of viral Fos (v-Fos), deficient in transcriptional repression function, does not induce apoptosis and v-Fos/p300 complexes were not detected in serum-free-treated cells.
Over-expression of p300 resulted in activation of apoptosis in both the hamster cells and in human tumor cells, but only in the cells expressing wild-type p53. In line with these observations, p300 has been shown to be an important component of p53-signaling, including p53-induced apoptosis.61,62,63,64 Over-expression of p300 has been shown to result in increased levels of p53.65 Therefore, we suggest the link between c-Fos and p53-induced apoptosis is p300. The primary mechanism, by which c-Fos:p300 complexes effect p53-induced apoptosis, is not clear, however, our results indicate that it is not a sequestering phenomenon. Unexpectedly, over-expression of the C-terminal domain of p300 (aa 1572–2370), a region that interacts with c-Fos and/or p53, did not block c-Fos-induced apoptosis. Instead, apoptosis was enhanced, implying that this is a functionally active complex. Since this p300-C terminal fragment contains a considerable amount of the C-terminal transactivation domain of p300, it is conceivable that it could participate in p53- and Fos-dependent gene regulation. We found that over-expression of the N-terminal fragment (aa 1–596) blocked death and there was no effect when the middle portion was expressed (aa 744–1571). Coincident with rescue from cell death, over-expression of the N-terminal fragment resulted in reduced expression of p300 protein. Because over-expression of p300 results in cell death, reduced levels could account for the observed rescuing effect. We have subcloned the p300 fragments into a tetracycline inducible system and examined the effects of expression in rat mesangial cells, when treated with low serum or H2O2. We found the same protective effect of the N-terminal fragment and an apoptosis-inducing effect of the C-terminal fragment (manuscript in review).
Published reports have shown that co-transfection of p53 with a C-terminal fragment of p300 (aa 1514–1922), lacking a portion of the C-terminal transactivation domain, inhibited p53-induced apoptosis by ∼50%.62 This data is not at odds with our observations, since this fragment does not contain a transactivation domain and could therefore sequester p53 as a non-functional complex. The current observation actually suggests that the C-terminal p53/Fos binding and the C-terminal transactivation domain of p300 are both necessary and sufficient for p300-mediated apoptosis. Further, this data would imply that the c-Fos, p53 and p300 promote apoptosis through transcriptional mechanisms.
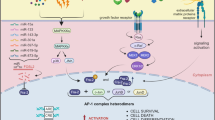
Because p300/CBP represents a common factor, required for function of nuclear receptors, CREB and AP-1, it has been suggested to be an integrator of multiple signal transduction pathways.40 The number of signal transduction pathways that require the activity of p300 is so large that the availability of these proteins can be readily envisioned as a limiting factor for the appropriate execution of many biological processes, if not for the maintenance of homeostasis.66 That is to say that these pathways may antagonize each other by virtue of their convergence on a common coactivator. This idea could be extended to provide a possible mechanism of how a cell might sense ‘mixed signals,’ one of the first proposals put forth as a cell signaling mechanism responsible for apoptosis. Because raising the levels of CBP can abolish the antagonistic effects of nuclear receptors and AP-1, regulation of CBP expression is likely to be of critical importance in determining the transcriptional consequences of simultaneous activation of multiple pathways. The model proposed in Figure 8 keeps into account the following observations: (i) when preneoplastic cells are placed in anti-proliferative conditions, they undergo apoptosis rather than growth arrest;1 (ii) the p53-MDM2-p300 complex is subjected to strict regulation in response to signals, which require the transcriptional activity of p53;66 (iii) over-expression of transcription factor, E2F1, which binds p300 in the C-terminal region, results in decreased MDM2 expression and apoptosis;67 (iv) the C-terminal domain of p300, p53 and c-Fos appear to be sufficient to induce apoptosis. Based on these data, we envision that specific apoptotic signal transduction pathways invoke the c-Fos:p300 complex through post-translational modification of c-Fos. p53 would also be modified and would simultaneously bind to this region. It is quite possible that this could result in accumulation of p53, due to repression of the MDM-2 gene or due to inhibition of MDM-2:p53 interactions through constraints on the accessibility. This interpretation must be regarded as conditional and future work is needed to determine whether these events constitute the signaling pathways that mediate other transcription factor-induced apoptosis.

A schematic model of the regulation of the p53-dependent apoptotic activity of c-Fos by p300
Cellular changes required for neoplastic transformation usually involve both deregulation of apoptosis and cell proliferation. p300 gene mutations have been detected in carcinomas of the digestive tract.55 In addition, a translocation in the p300 gene has been detected in acute myeloid leukemia,54 suggesting that p300 acts as a negative regulator of cell growth.68 While mutated forms of the p300/CBP facilitate uncontrolled proliferation, the normal p300 function is essential for survival as demonstrated by data showing that p300−/− mice die in utero.49 The involvement of p300 in human disease, coupled with biochemical evidence indicating that it is essential in cell-cycle control, cellular differentiation and human development, makes it likely that it is a tumor suppressor protein.68 In this regard, p300 could function as a general mediator of transcription factor-induced apoptosis.
Materials and Methods
Cell culture
SHE cell lines were established as described previously.69,70 The preneoplastic, hamster cell lines, sup+I cells and sup−II cells, used in this work have also been described.1 Development of the Sup−II/FosER and Sup−II/v-Fos has been described earlier.2 RKO human colorectal carcinoma cells were obtained from Dr Bert Vogelstein, Johns Hopkins University. HT1080p53wt human fibrosarcoma cells were purchased from ATTC. HT1080p53mutant cells were received from Dr Bernard Weissman, UNC, Chapel Hill. All cells were maintained in Dulbecco's modified IBR medium (GIBCO, Grand Island, NY, USA) supplemented with 10% fetal bovine serum (Hyclone), 100 U/ml penicillin, and 100 μg/ml streptomycin and were incubated at 37°C in 10% CO2 in air.
Antibodies
The p300 antibody was the anti-human p300, Ct-Power Clonal (mixed mouse monoclonal IgG1) purchased from Upstate Biotechnology. The rabbit polyclonal c-Fos (SC-52) and cyclin D1 antibodies were purchased from Santa Cruz.
Analysis of c-Fos:p300 immune complexes
Sup+I, sup−II, sup−II /FosER and Sup−II/v-Fos cells were plated (1.5×106/150 mm dish) 24 h in 10% FBS phenol red free medium. To induce apoptosis, sup+I and sup−II cells were washed twice in warm calcium- and magnesium-free phosphate-buffered saline (CMF-PBS) and placed in medium plus 0.1% FBS. Sup−II /FosER cells were rinsed twice with CMF-PBS and placed in serum-free medium with or without 1 μM estradiol (E2) (Sigma). After specified incubation times, cells were lysed in modified NP-40 lysis buffer (25 mM HEPES (pH 7.5), 12.5 mM MgCl2, 150 mM KCl, 0.5% NP-40, 1 mM dithiothreitol) containing 10 ug of leupeptin per ml, 1% aprotinin, and 0.5 mM phenylmethylsulfonyl fluoride. Glycerol was added to a final concentration of 8%. After incubation (1 h at 4°), extracts were centrifuged at 14 000 r.p.m. for 30 min and supernatants were collected. Extracts were precleared with an excess of Protein A-sepharose. Typically, one half of the precleared extract was immunoprecipitated with the anti-p300 antibody (1 : 100) and the remaining with the anti-c-Fos antibody (1 : 100). Immunoprecipitations were incubated overnight at 4°. Immune complexes were retrieved with protein A, washed 3–5 times in lysis buffer, and eluted in 2× Lammeli sample buffer. Samples were analyzed by Western blot.
Transfections
Cells were trypsinized and plated 24 h prior to transfection. CMVβ P300, CMVβ P300 CHA, CMVβ β-galactosidase, CMVβ 5.3ATGdel33, GST-C-term p300, and GST-N-term p300 vectors were a gift from Dr Richard Eckner, Dana-Farber Cancer Institute. GST-Middle p300 was a gift from Dr Yang Shi, Dana-Farber Cancer Institute. DNAs (10 μg/ml) were transfected into sup+I, sup−II, sup−II /FosER, HT1080 and RKO cells (2×106/0.8 ml) by electroporation. Electroporations were done in serum-free medium with 300 volts at a capacitance of 960 μF (Gene Pulser, BRL).
Analysis and quantitation of apoptosis
DNA fragmentation was analyzed on combined attached and detached cells. Cells were collected, rinsed with CMF-PBS, lysed in 50 μl volumes of lysis buffer (10 mM EDTA, 50 mM Tris, pH 8.0, 0.5% sodium lauryl sarcosine, 0.5 mg/ml proteinase K) and then incubated at 50°C for 1 h. RNase A (0.5 mg/ml) was added and lysates were incubated for an additional h. DNA was electrophoresed in a 1% agarose gel in 0.5× TBE running buffer (0.05 M Tris base, 0.05 M boric acid, 1 mM disodium EDTA) for 1 h at 90 volts.
Apoptosis was quantitated by FACScan analysis of TUNEL-labeled cells, using dUTP:FITC and terminal transferase (TdT) from Boehringer Mannheim. Briefly, the cell pellets were washed with cold PBS and fixed with 1% paraformaldehyde in PBS for 15 min. The pellet was washed in PBS and the cells were then fixed with 70% ethanol. The cells were rinsed in PBS plus 1% BSA and the labeling reaction was performed. Each cell sample was divided into two. One half was used to determine background fluorescence by omitting the terminal transferase enzyme (TdT) from the reaction mixture. The TdT was added to the second half and increases in fluorescence (FL1) were evaluated by FACScan analysis. Clumps and doublets were excluded from the analysis by using forward scatter vs propidium iodide fluorescence (FL2-W).
Protein extraction and immunoblotting
Cells were rinsed twice in cold PBS. A volume of sodium dodecyl sulfate (SDS)-polyacrylamide gel Laemmli sample buffer (100°C) was added directly to cells to achieve a final concentration of 3×106 cells/ml. Cell lysates were collected, boiled for 5 min, and sonicated. Aliquots equal to 2×105 cells were subjected to SDS-polyacrylamide gel electrophoresis at 150 V for 1 h and transferred to nitrocellulose at 100 V for 1 h in transfer buffer containing 19.1 mM Tris-HCl, 190 mM glycine.
Abbreviations
- AP-1:
-
activating protein-1
- CBP:
-
CREB-binding protein
- E2:
-
estradiol
- sup+I:
-
tumor suppressor gene plus preneoplastic stage I
- sup−II:
-
tumor suppressor gene minus preneoplastic stage II
- TdT:
-
terminal transferase
References
Preston GA, Lang JE, Maronpot RR and Barrett JC . (1994) Regulation of apoptosis by low serum in cells of different stages of neoplastic progression: enhanced susceptibility after loss of a senescence gene and decreased susceptibility after loss of a tumor suppressor gene. Cancer Res. 54: 4214–4223
Preston GA, Lyon TT, Yin Y, Lang JE, Solomon G, Annab L, Srinivasan DG, Alcorta DA and Barrett JC . (1996) Induction of apoptosis by c-Fos protein. Mol. Cell Biol. 16: 211–218
Afshari CA, Bivins HM and Barrett JC . (1994) Utilization of a fos-lacZ plasmid to investigate the activation of c-fos during cellular senescence and okadaic acid-induced apoptosis. J. Gerontol. 49: B263–269
Coletta F, Polentarutti N, Sironi M and Mantovani A . (1992) Expression and involvement of c-fos and c-jun protooncogenes in programmed cell death induced by growth factor deprivation in lymphoid cell lines. J. Biol. Chem. 267: 18278–18283
Day ML, Zhao X, Wu S, Swanson PE and Humphrey PA . (1994) Phorbol ester-induced apoptosis is accompanied by NGFI-A and c-fos activation in androgen-sensitive prostate cancer cells. Cell Growth Differ. 5: 735–741
Loebermann DA, Gregory B and Hoffman B . (1998) AP-1 (Fos/Jun) transcription factors in hematopoietic differentiation and apoptosis. Int. J. Oncol. 12: 685–700
Marti A, Jehn B, Costello E, Keon N, Ke G, Martin F and Jaggi R . (1994) Protein kinaseA and AP-1 (c-Fos/JunD) are induced during apoptosis of mouse mammary epithelial cells. Oncogene 9: 1213–1223
Matsuoka M, Wispriyono B and Igisu H . (1997) Induction of c-fos gene by mercury chloride in LLC-PKI cells. Chem. Biol. Interact. 108: 95–106
Mils V, Piette J, Barette C, Veyrune J, Tesniere A, Escot C, Guilhou J and Basset-Seguin N . (1997) The proto-oncogene c-fos increases the sensitivity of keratinocytes to apoptosis. Oncogene 14: 1555–1561
Rich KA, Zhan Y and Blanks JC . (1997) Aberrant Expression of c-Fos accompanies photoreceptorcell death in the rd mouse. J. Neuro. Biol. 3216: 593–612
Schadendorf D, Kern MA, Artuc M, Pahl HL, Rosenbach T, Fichtner I, Nurnberg W, Stuting S, von Stebut E, Worm M, Makki A, Jurgovsky K, Kolde G and Henz BM . (1996) Treatment of melanoma cells with the synthetic retinoid CD437 induces apoptosis via activation of AP-1 in vitro, and causes growth inhibition in xenografts in vivo. J. Cell Biol. 135: 1889–1898
Schaerli P and Jaggi R . (1998) EGF-induced programmed cell dath of human mammary carcinoma MDA-MB-468 cells is preceded by activation of AP-1. Cell. Mol. Life Sci. 54: 129–138
Bies J and Wolfe L . (1995) Acceleration of apoptosis in transforming growth factor beta 1-treated M1 cells ectopically expressing B-Myb. Cancer Res. 55: 501–504
Abbadie C, Kabrun N, Bouali F, Smardova J, Stehelin D, Vandenbunder B and Enrietto PJ . (1993) High levels of c-rel expression are associated with programmed cell death in the developing avian embryo and in bone marrow cells in vitro. Cell. 75: 899–912
Beg AA and Baltimore D . (1996) An essential role for NF-κB in preventing TNF-alpha-induced cell death. Science. 274: 782–784
Ferrer I, Olive M, Blanco R, Cinos C and Planas AM . (1996) Selective c-Jun over-expression is associated with ionizing radiation-induced apoptosis in the developing cerebellum of the rat. Brain Res. Mol. Brain Res. 38: 91–100
Ferrer I, Segui J and Olive M . (1996) Strong c-Jun immunoreactivity is associated with apoptotic cell death in human tumors of the central nervous system. Neurosci. Lett. 214: 49–52
Lee CW, Sorensen TS, Shikama N and LaThangue NB . (1998) Functional interplay between p53 and E2F through co-activator p300. Oncogene 16: 2695–2710
Liu C, Rnagnekar VM, Adamson E and Mercola D . (1998) Suppression of growth and transformation and induction of apoptosis by EGR-1. Cancer Gene Ther. 5: 3–28
Muthukkumar S, Prakash N, Sells SF, Maddiwar NG, Jacob RJ and Rangnekar VM . (1995) Role of EGR-1 in thapsigargin-inducible apoptosis in the melanoma cell line A375-C6. Mol. Cell. Biol. 15 6262–6272
Sala A, Casslla I, Grasso L, Bellon T, Reed JC, Miyashita T and Peschle C . (1996) Apoptotic response to oncogenic stimuli: cooperative and antagonistic interactions between c-myb and the growth suppressor c-myb. Cancer Res. 56: 1991–1996
Shi Y, Glynn JM, Guilbert LJ, Cotter TG, Bissonnette RP and Green DR . (1992) Role for c-myc in activation-induced apoptotic cell death in T cell hybridomas. Science 257: 212–214
Skov S, Nielsen M, Bregenholt S, Odum N and Claesson MH . (1998) Activation of stat-3 is involved in the induction of apoptosis after ligation of major histocompatibility complex class I molecules on human Jurkat T cells. Blood. 91: 3566–3573
Solomoni P, Perrotti D, Martinez R, Franceschi C and Calabretta B . (1997) Resistance to apoptosis in CTLL-2 cells constitutively expressing c-Myb is associated with induction of BCL-2 expresion and Myb-dependent regulation of bcl-2 promoter activity. Proc. Natl. Acad. Sci. USA 94: 3296–3301
Weston E . (1998) Myb proteins in life, death, and differentiation. Curr. Opin. Genet. Dev. 8: 76–81
Yamada T, Yamaoka S, Goto T, Nakai M, Tsujimoto Y and Hatanaka M . (1994) The human T-cell leukemia virus type I Tax protein induces apoptosis which is blocked by the BCL-2 protein. J. Virol. 68: 3374–3379
Caelles C, Heimberg A and Karin M . (1994) p53-dependent apoptosis in the absence of transcriptional activation of p53-target genes. Nature 370: 220–223
Hermeking H and Eick D . (1994) Mediation of c-myc-induced apoptosis by p53. Science 265: 2091–2093
Ledda-Columbano G M, Coni P, Faa G, Manenti G and Columbano A . (1992) Rapid induction of apoptosis in rat liver by cycloheximide. Amer. J. Path. 140: 545–549
Piedrafita FJ and Pfahl M . (1997) Retinoid-induced apoptosis and Sp1 cleavage occur independently of transcription and require caspase activation. Mol. Cell. Biol. 17: 6348–6358
Waring P . (1990) DNA fragmentation induced by macrophages by gliotoxin does not require protein synthesis and is preceded by raised inositol triphosphate levels. J. Biol. Chem. 265: 14476–14480
Wyllie AH, Kerr JFR and Currie AR . (1980) Cell death: The significance of apoptosis. Int. Rev. Cytol. 68: 251–306
Arany Z, Huang LE, Eckner R, Bhattacharya S, Jiang C, Goldberg MA, Bunn HF and Livingston DM . (1996) An essential role for p300/CBP in the cellular response to hypoxia. Proc. Natl. Acad. Sci. USA 93: 12969–12973
Bannister AJ and Kouzarides T . (1995) CBP-induced stimulation of c-Fos activity is abrogated by E1A. EMBO J. 14: 4758–4762
Bex F, Yin MJ, Burny A and Gaynor RB . (1998) Differential transcriptional activation by human T-cell leukemia virus type 1 Tax mutants is mediated by distinct interactions with CREB binding protein and p300. Mol. Cell. Biol. 18: 2392–2405
Bhattacharya S, Eckner R, Grossman S, Oldread E, Arany Z, D'Andrea A and Livingston DM . (1996) Cooperation of Stat2 and p300/CBP in signalling induced by interferon-alpha. Nature. 383: 344–347
Facchinetti V, Loffarelli L, Schreek S, Oelgeschlager M, Luscher B, Introna M and Golay J . (1997) Regulatory domains of the A-Myb transcription factor and its interaction with the CBP\p300 adaptor molecules. Biochem. J. 324: 729–736
Gerritsen ME, Williams AJ, Neish AS, Moore S, Shi Y and Collins T . (1997) CREB-binding protein/p300 are transcriptional coactivators of p65. Proc. Natl. Acad. Sci. USA 94: 2927–2932
Horvai AE, Xu L, Korzus E, Brard G, Kalafus D, Mullen TM, Rose DW, Rosenfeld MG and Glass CK . (1997) Nuclear integration of JAK/STAT and Ras/AP-1 signaling by CBP and p300. Proc. Natl. Acad. Sci. USA 94: 1074–1079
Kamei Y, Xu L, Heinzel T, Torchia J, Kurokawa R, Gloss B, Lin SC, Heyman RA, Rose DW, Glass CK and Rosenfeld MG . (1996) A CBP integrator complex mediates transcriptional activation and AP-1 inhibition by nuclear receptors. Cell. 85: 403–414
Perkins ND, Felzien LK, Betts JC, Leung K, Beach DH and Nabel GJ . (1997) Regulation of NF-kappaB by cyclin-dependent kinases associated with the p300 coactivator. Science 275: 523–527
Egan C, Jelsma TN, Howe JA, Bayley ST, Ferguson B and Branton PE . (1988) Mapping of cellular proteins-binding sites on the products of early-region 1A of human adenovirus type 5. Mol. Cell. Biol. 9: 3955–3959
Whyte P, Williamson NM and Harlow E . (1989) Cellular targets for transformation by the adenovirus E1A proteins. Cell. 56: 67–75
Arany Z, Newsome D, Oldread E, Livingston DM and Eckner R . (1995) A family of transcriptional adaptor proteins targeted by the E1A oncoprotein. Nature 374: 81–84
Eckner R, Arany Z, Ewen M, Sellers W and Livingston DM . (1994) The adenovirus E1A-associated 300-kD protein exhibits properties of a transcriptional coactivator and belongs to an evolutionarily conserved family. Cold Spring Harb. Symp. Quant. Biol. 59: 85–95
Eckner R, Ewen ME, Newsome D, Gerdes M, DeCaprio JA, Lawrence JB and Livingston DM . (1994) Molecular cloning and functional analysis of the adenovirus E1A-associated 300-kD protein (p300) reveals a protein with properties of a transcriptional adaptor. Genes Dev. 8: 869–884
Lundblad JR, Kwok RP, Laurance ME, Harter ML and Goodman RH . (1995) Adenoviral E1A-associated protein p300 as a functional homologue of the transcriptional co-activator CBP. Nature 374: 85–88
Kawasaki H, Eckner R, Yao TP, Taira K, Chiu R, Livingston DM and Yokoyama KK . (1998) Distinct roles of the co-activators p300 and CBP in retinoic-acid-induced F9-cell differentiation. Nature 393: 284–289
Yao T P, Oh SP, Fuchs M, Zhou ND, Ch'ng LE, Newsome D, Bronson RT, Li E, Livingston DM and Eckner R . (1998) Gene dosage-dependent embryonic development and proliferation defects in mice lacking the transcriiptional integrator p300. Cel. 93: 361–372
Liu Y and Kitsis RN . (1996) Induction of DNA synthesis and apoptosis in cardiac myocytes by E1A oncoprotein. J. Cell Biol. 133: 325–334
Rao L, Debbas M, Sabbatini P, Hockenbery D, Korsmeyer S and White E . (1992) The adenovirus E1A proteins induce apoptosis, which is inhibited by the E1B 19-kDa and Bcl-2 proteins. Proc. Natl. Acad. Sci. USA 89: 7742 7746
Moran E . (1993) DNA tumor virus transforming proteins and the cell cycle. Curr. Opin. Genet. Dev. 3: 63–70
Mymryk JS, Shire K and Bayley ST . (1994) Induction of apoptosis by adenovirus type 5 E1A in rat cells requires a proliferation block. Oncogene 9: 1187–1193
Muraoka M, Konishi M, Kikuchi Yanoshita R, Tanaka K, Shitara N, Chong JM, Iwama T and Miyaki M . (1996) p300 gene alterations in colorectal and gastric carcinomas. Oncogene 12: 1565–1569
Ida K, Kitaayashi I, Taki T, Taniwaki M, Noro K, Yamamoto M, Ohki M and Hayashi Y . (1997) Adenoviral E1A-associated protein p300 is involved in acute myeloid leukemia with t(11;22)(q23;q13). Blood 90: 4699–4704
Koi M, Afshari CA, Annab LA and Barrett JC . (1989) Role of a tumor-suppressor gene in the negative control of anchorage-independent growth of Syrian hamster cells. Proc. Natl. Acad. Sci. USA 86: 8773–8777
Owen RD, Hosoi J, Montgomery JC, Wiseman R and Barrett JC . (1993) Coordinate regulation of collagen II (alphaI) and H19 expression in immortalized hamster cells. Cell Growth and Differ. 4: 1013–1021
Wiseman RW, Montgomery JC, Hosoi J, Hou EW, Cochran CJ, Lamb PW and Barrett JC . (1991) Identification of genes associated with tumor suppresion in Syrian hamster embryo cells. Environ. Health Perspect. 93: 105 109
Lee JS, See RH, Deng T and Shi Y . (1996) Adenovirus E1A downregulates cJun- and JunB-mediated transcription by targeting their coactivator p300. Mol. Cell Biol. 16: 4312–4326
Smits PHM, de Wit L, van der Eb AJ and Zantema A . (1996) The Adenovirus E1A-associated 300kDa Adaptor Protein Counteracts the Inhibition of the Collagenase Promoter by E1A and Represses Transformation. Oncogene 12: 1529–1535
Lill NL, Grossman SR, Ginsberg D, DeCaprio J and Livingston DM . (1997) Binding and modulation of p53 by p300/CBP coactivators. Nature 387: 823–826
Avantaggiati ML, Ogryzko V, Gardner K, Giordano A, Levine AS and Kelly K . (1997) Recruitment of p300/CBP in p53-dependent signal pathways. Cell 89: 1175–1184
Lee JS, Galvin KM, See RH, Eckner R., Livingston D, Moran E and Shi Y . (1995) Relief of YY1 transcriptional repression by adenovirus E1A is mediated by E1A-associated protein p300 [published erratum appears in Genes Dev 1995 Aug 1;9(15):1948-9]. Genes Dev. 9: 1188–1198
Scolnick DM, Chehab NH, Stavridi ES, Lien MC, Caruso L, Moran E, Berger SL and Halazonetis TD . (1997) CREB-Binding protein and p300/CBP-associated factor are transcriptional coactivators of the p53 tumor suppressor protein. Cancer Res. 57: 3693–3696
Yuan Z-M, Huang Y, Ishiko T, Nakada S, Utsugisawa T, Shioya H, Utsugisawa Y, Yokoyama K, Weichselbaum R, Shi Y and Kufe D . (1999) Role for p300 in stabilization of p53 in the response to DNA damage. J. Biol. Chem. 274: 1883–1886
Giordano A and Avantaggiati ML . (1999) p300 and CBP: Partners for life and death. J. Cell. Phys. 181: 218 230
Yang HL, Dong YB, Elliott MJ, Liu TJ, Atienza Jr, C, Stilwell A and McMasters KM . (1999) Adenovirus-mediated E2F-1 gene transfer inhibits MDM2 expression and efficiency induces apoptosis in MDM2-over-expressing tumor cells. Clin. Can. Res. 5: 2242–2250
Giles RH, Peters DJ and Breuning MH . (1998) Conjunction dysfunction: CBP/p300 in human disease. Trends Genet. 14: 178–183
Koi M and Barrett JC . (1986) Loss of tumor-suppressive function during chemically induced neoplastic progression of Syrian hamster embryo cells. Proc. Natl. Acad. Sci. USA 83: 5992–5996
Oshimura M, Hesterberg WW and Barrett JC . (1986) An early, non-random karyotypic change in immortal Syrian hamster cell lines transformed by asbestos: trisomy of chromosome 11. Cancer Genet. Cytogenet. 22: 225–237
Acknowledgements
We thank Drs Cindy Afshari, Rick Paules, and Mike Colman for serving as internal reviewers and for their critical comments of the manuscript. We also thank Drs Richar Eckner, Julian Preston, Ronald Falk, and Marten Spegelmark for their helpful advice and discussions. We thank Drs R Eckner and Yang Shi for the generous gifts of plasmids. The authors thank Dr Ronald Falk for generously providing the opportunity for the studies to be completed in his laboratory.
Author information
Authors and Affiliations
Corresponding author
Additional information
Edited by C Thiele
Rights and permissions
About this article
Cite this article
Preston, G., Srinivasan, D. & Barrett, J. Apoptotic response to growth factor deprivation involves cooperative interactions between c-Fos and p300. Cell Death Differ 7, 215–226 (2000). https://doi.org/10.1038/sj.cdd.4400637
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.cdd.4400637
