
Abstract
Methylation of tumor suppressor genes has been implicated in breast cancer development. However, methylation profiles of different breast lesions, subtypes of carcinoma in particular, have not been examined in detail. In this study, we use methylation-specific PCR (MSP) to generate gene methylation profiles of different breast lesions and to test the clinical utility of such profiles. We examined the methylation status of three genes, RARβ2, RASSF1A, and cyclin D2, on 102 samples of breast tissue, from benign (n = 36), to in situ carcinoma (n = 21), to invasive carcinoma (n = 45). We found that almost all cases of invasive carcinoma (96%) contained at least one methylated gene from our panel, whereas gene methylation was less common among benign lesions (42%) and in situ carcinoma (76%). Of the three genes, cyclin D2 methylation was most specific for malignancy because only 1 of 35 benign cases was methylated at this gene (1 case was not informative). The major histologic subtypes of invasive carcinoma show similar methylation profiles in the genes examined. We next performed MSP analysis on archival breast fine-needle aspiration (FNA) biopsy samples and corresponding surgical biopsy specimens and found a high concordance between the two types of specimens. We then analyzed 17 breast FNA biopsy samples with an indeterminate diagnosis. In this setting, MSP had a high specificity (100%) and modest sensitivity (67%) for identifying malignancy.
Similar content being viewed by others


Main
Mutations in tumor suppressor genes, such as BRCA1 and p53, are known to be important in breast cancer development. Epigenetic events, such as DNA methylation, may also play an important role. DNA methylation, the addition of a methyl group to the cytosine residue of CpG dinucleotides within gene promoters, has been implicated in cancer development because many tumor suppressor genes are silenced by DNA methylation (1). Genes found to be methylated in breast cancers include the following: p16, cyclin D2, BRCA1, ER, PR, 14–3-3σ, E-cadherin, TIMP-3, GSTP1, RASSF1A, Twist, and RARβ2 (2, 3, 4, 5, 6, 7, 8, 9). The reported frequency of methylation of these genes in breast carcinoma ranges from 15% for BRCA1 to >90% for 14–3-3σ.
RARβ2 is one of the nuclear receptors for the active derivative of vitamin A, retinoid acid (RA), which is critical for normal development and differentiation. Studies have shown that RARβ2 also mediates the anticancer effect of RA (10, 11, 12). Down-regulation of RARβ2 has been observed in a number of different types of human malignancy, including breast carcinoma (10, 13, 14, 15). In these cases, DNA methylation is responsible for the decreased transcription of RARβ2 more often than RARβ2 mutation (14, 16). The methylation of RARβ also confers RA resistance by facilitating the deacetylation of histones in the promoter region of RARβ (17). RASSF1A, the human Ras association domain family 1A gene, has also been implicated in multiple cancers, including lung, breast, and bladder carcinoma (18, 19, 20, 21, 22). Although its exact role in tumorigenesis is not certain, exogeneous expression of RASSF1A in tissue culture cells reduces in vitro colony formation and in vivo tumorigenecity (19). Increased expression of cyclin D2, a D-type cyclin, has been associated with proliferation of tumor cells and poor prognosis in gastric carcinoma (23). However, down-regulation of cyclin D2 expression was noted in breast cancer cell lines, as well as primary tumors, in comparison to normal breast epithelial or stromal cells (4). This paradoxical down-regulation of cyclin D2 suggests other functions for this molecule, possibly in apoptosis (4).
Although methylation of individual tumor suppressor genes has been demonstrated in breast tumor development, there has not been a detailed characterization of the methylation profiles of breast carcinoma subtypes. Also, the utility of gene methylation profiling in diagnostic breast cytopathology has only begun to be addressed (24). Therefore, we generated methylation profiles of RARβ2, RASSF1A, and cyclin D2 from a spectrum of breast lesions ranging from benign to invasive to assess the clinical utility of such profiling in surgical and cytological specimens and to characterize different histologic subtypes of breast carcinoma.
MATERIALS AND METHODS
Case Selection
Cases were selected from Johns Hopkins Hospital archives after obtaining IRB approval. Thirty-six benign lesions (intraductal papillomas [n = 32], radial scar/sclerosing adenosis [n = 4]), 21 carcinomas in situ (ductal carcinoma in situ [n = 20], lobular carcinoma in situ [n = 1]), and 45 invasive carcinomas (ductal [n = 15], lobular [n = 15], tubular [n = 12], medullary [n = 1], papillary [n = 1], and carcinosarcoma [n = 1]) were included in the study. Eight cases of invasive carcinoma with corresponding FNA cytology and surgical specimens were also analyzed. An additional 17 indeterminate cytology specimens were analyzed, with each having a cytological diagnosis of “suspicious for malignancy,” “malignancy cannot be excluded,” or “atypia.”
DNA Extraction and Sodium Bisulfite Treatment
The procedures were carried out according to protocols described elsewhere (25). Briefly, paraffin-embedded tissue blocks were retrieved, and for each surgical pathology specimen, one section was prepared for H&E stain and morphologic evaluation, and four sections (10 μm thickness) were cut for DNA extraction. Paraffin was removed by incubating in 500 μL of xylene for 30 min at room temperature, twice, followed by two washes with 1 mL of 95% ethanol. The deparaffinized tissue sections were then allowed to dry and were incubated in 200 μL of 1 × TNES buffer (10 mm Tris, pH 8.0; 150 mm NaCl; 2 mm EDTA; 0.5% SDS; with 0.5 mg/mL proteinase K, at 55° C overnight. DNA was purified using a DNA Wizard Clean-up Kit according to the manufacturer's protocol (Promega, Madison, WI) and quantified. For archival cytological specimens, coverslips were removed from either air-dried, Diff-Quick–stained slides or from 95% ethanol-fixed, Papanicolaou-stained slides by immersion in xylene, then allowed to dry at room temperature. Next, 150 μL of 1 × TNES buffer was added to the dried surface of slides, and cells were removed by scraping the surface with a blade and transferring the material into an Eppendorf tube. Proteinase K was added to a concentration of 0.5 mg/mL and incubated at 55° C overnight. The supernatant was used for sodium bisulfite treatment and methylation-specific PCR (MSP) analysis according to published procedures (25). Briefly, 50 μL of DNA (≤1 μg) was denatured by adding 5.5 μL of 2 m NaOH for 10 min at 37° C. Next, 30 μL of 10 mm hydroquinone (Sigma) and 520 μL of 3 m sodium bisulfite (Sigma) at pH 5, both prepared fresh, were added. Samples were then layered with mineral oil and incubated at 50° C overnight. Modified DNA was then purified using the DNA Wizard Clean-Up Kit (Promega) according to the manufacturer’s protocol and eluted with 50 μL of water. Chemical modification was completed by treating DNA with 5.5 μL of 3 m NaOH and incubating for 5 min at room temperature. DNA was precipitated with ethanol and resuspended in 20 μL of water and stored at −20° C until use.
Methylation-Specific PCR
The RARβ2 and cyclin D2 sequences of primers used in this study have been reported previously (24; Invitrogen, Carlsbad, CA). The RASSF1A primer sequences were as follows: unmethylated forward, 5′-GGTTGTATTTGGTTGGAGTG-3′; unmethylated reverse, 5′-CTACAAACCTTTACACACAACA-3′; methylated forward, 5′-AGCGAAGTACGGGTTTAATC-3′; and methylated reverse, 5′-GCACCACGTATACGTAACG-3′. The PCR mixture contained 1 × MSP buffer (17 mm ammonium sulfate; 67 mm Tris, pH 8.8; 67 mm MgCl2; and 10 mm 2-mercaptoethanol), dNTPs (each at 1.25 mm), primers (100 ng/mL), and sodium bisulfite-modified DNA (1.5 μL) in a final volume of 50 μL. Reactions were hot-started at 95° C for 5 min before the addition of 10 μL of 1:10 Taq polymerase (RedTaq; Sigma, St. Louis, MO) or were performed without hot start when using Jump-start Red-Taq (Sigma). Amplification was carried out in a Hybaid Omnigene thermal cycler for 36 cycles (30 s at 95° C, 30 s at 56° C, and 45 s at 72° C), followed by a 5-min extension at 72° C. A negative control (water only) and positive controls were included for each set of MSP. DNA from the breast cancer cell line MDA-MB-231, was used as a positive control for methylated products, and DNA from normal white blood cells or the EBV-transformed lymphoblast cell line, NLBL1, was used as a positive control for unmethylated reactions. Each PCR reaction was then subjected to electrophoresis on a 2% agarose gel and stained with ethidium bromide to visualize PCR products.
Statistical Analysis
All p-values are based on Pearson Chi-square test except the ones with asterisks, which are based on a Fisher’s Exact test (because of sparse data).
RESULTS
Promoter Methylation Frequency in Benign Breast Lesions, In Situ Carcinomas, and Invasive Carcinomas
We analyzed the methylation status of the RARβ2, RASSF1A, and cyclin D2 gene promoters in 36 archival samples of benign breast lesions, predominantly intraductal papillomas (Fig. 1). At least one of the three promoters was methylated in 42% of the cases (cumulative methylation index, CMI). This was predominantly due to methylation of RARβ2 and RASSF1A, which displayed a much higher methylation frequency (33% and 34%, respectively) than cyclin D2 (<3%). There was no difference between the mean ages of the patients in the groups with methylated genes versus unmethylated genes (55.6 y and 57.7 y, respectively). We next examined the methylation frequency of the same gene promoters in a group of 21 in situ breast carcinomas, predominantly ductal carcinoma in situ (Fig. 1). This in situ carcinoma group had a significantly higher frequency of promoter methylation than the benign lesions (CMI of 76% and 42%, respectively, P = .03). The methylation frequency for the individual genes, RARβ2, RASSF1A, and cyclin D2, was 60%, 62%, and 57%, respectively, among these in situ carcinomas. There was a significant difference in the methylation frequency of cyclin D2 between benign and in situ carcinomas (57% versus 3%, P < .001*). There was also a trend toward more frequent methylation of the other two genes among in situ carcinomas, relative to benign lesions, but these differences were not statistically significant (RARβ2: 60% versus 33%, P = .06; RASSF1A: 62% versus 34%, P = .05).

Frequency of gene methylation in benign breast lesions, in situ carcinomas, and invasive carcinomas. The percentage of cases that contain methylated RARβ2, RASSF1A, and cyclin D2 genes is indicated. The percentage of cases that contain at least one methylated gene from this three-gene panel (cumulative methylation index, CMI) is also indicated.
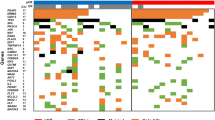
The methylation frequency of the same gene promoters was then analyzed in a group of 45 invasive breast carcinomas (Fig. 1). This group included the following subtypes of invasive carcinoma: ductal carcinoma, lobular carcinoma, tubular carcinoma, and several less common carcinomas. The cumulative methylation index for these invasive carcinomas was 96%. The individual genes (RARβ2, RASSF1A, and cyclin D2) were methylated in 64%, 64%, and 71% of the cases, respectively. The cumulative methylation index of this group is significantly higher than that of the benign group (96% versus 42%, P < .0001) or the in situ carcinoma group (96% versus 76%, P = 0.01*). There was no significant difference in the methylation profiles among the subtypes of invasive carcinoma using this three-gene panel (P > 0.05; Fig. 2).

Gene methylation profiles of histologic subtypes of invasive breast carcinomas. Each row represents MSP results from an individual case. A filled box indicates methylation of that gene, whereas an open box indicates no methylation; ni indicates cases that were not informative because of the lack of a product upon MSP.
High Concordance of Methylation Profiles between Corresponding Surgical and Cytological Specimens
Eight cases of invasive carcinoma with corresponding cytology and surgical specimens were analyzed to determine the feasibility of MSP analysis on archival cytological smears and the concordance of methylation profiles between the two types of specimens. Preliminary studies in our laboratory using air-dried, Diff-Quik–stained tissue culture cell smears and ethanol-fixed, Papanicolaou-stained smears indicated that the two cytological preparations yielded equally informative MSP results (data not shown), and thus cytological archival material was suitable for this analysis. We then compared the methylation profiles from archival cytological specimens with those of the corresponding surgical specimens. The concordance of RARβ2, RASSF1A, and cyclin D2 methylation was 63%, 83%, and 100%, respectively, between cytology and surgical specimens (Fig. 3). The cyclin D2 methylation profile matched exactly between surgical and cytology specimens, whereas the RASSF1A MSP results differed in one case. RARβ2 displayed more discordance in MSP results between cytologic and surgical specimens, possibly because of sampling differences between specimens or differences in the sensitivity of the MSP assays for different genes.

Correlation of methylation profiles between corresponding surgical and cytologic biopsies. Each row represents MSP results from an individual patient. The Cyto column contains results from FNA samples, and the SP column contains the results from the corresponding surgical pathology biopsies. A filled box indicates methylation of that gene, whereas an open box indicates no methylation; ni indicates cases that were not informative because of the lack of a product upon MSP.
Methylation Profiles of Atypical Breast FNA Biopsies
To test the utility of MSP to identify malignancy in atypical breast FNA biopsies, we examined the methylation profiles of 17 atypical breast FNA samples and compared these results to the ultimate histologic diagnosis. These histologic diagnoses included 12 cases of malignancy (3 DCIS and 9 invasive carcinomas) and 5 benign cases. Of the 17 FNAs, 8 cases contained at least one methylated gene from our three-gene profile (Fig. 4). All of the eight FNA cases that contained at least one methylated gene were ultimately found to be malignant. Conversely, none of the benign lesions were found to contain any methylated genes in the FNA sample. However, four malignant cases did not contain any methylated genes from our three-gene panel. Consequently, the sensitivity and specificity of this assay for malignancy in these atypical breast FNAs is 67% and 100%, respectively.

Gene methylation profiles on atypical breast FNA specimens. Each row represents MSP results from an individual FNA sample that was originally classified as atypical. Cases 1–5 are FNA samples from lesions that were subsequently found to be benign, Cases 6–8 are from DCIS, and Cases 9–17 are from invasive carcinomas. A filled box indicates methylation of that gene, whereas an open box indicates no methylation; ni indicates cases that were not informative because of the lack of a product upon MSP.
DISCUSSION
Gene methylation is a process in which the enzyme DNA methyltransferase adds a methyl group to the cytosine on CpG dinucleotides within the promoter of a gene. DNA methylation represses gene expression by attracting methyl domain–binding proteins to the region, which recruit other transcriptional repressors to the promoter. Binding of these proteins also influences the posttranslational modification of histones, which further represses transcription (reveiwed in 26).
RARβ2 methylation has been examined previously in breast cancer cell lines and primary tumors. In these studies, the frequency of RARβ2 methylation ranged from 38–63% (10, 16, 17) in invasive carcinomas, whereas the gene was rarely methylated in benign breast tissue. Our results are similar but suggest that RARβ2 methylation might be slightly more frequent in our patients. We have also found that RARβ2 methylation is a relatively frequent event in benign intraductal papillomas and DCIS. We also have extended this work by examining the most common histologic subtypes of invasive breast carcinomas. We did not find a statistically significant difference in the methylation frequency of RARβ2 in ductal, tubular, or lobular breast carcinomas, suggesting that RARβ2 methyation may play a similar role in the carcinogenic pathways used by these three subtypes of breast carcinomas and is likely to be an early event.
RASSF1A methylation has also been previously described in primary breast carcinomas and breast carcinoma cell lines. In general, the frequency of RASSF1A methylation in invasive breast carcinomas is high (49–65%) (27, 28, 29), but it was significantly lower in one study (9%) (30). Our data agree with studies suggesting that RASSF1A methylation is a relatively frequent event in invasive breast carcinoma. Our data found somewhat different frequencies of RASSF1A methylation among three common histologic subtypes of breast carcinoma, but these differences were not statistically significant. Another study found that 42% of DCIS lesions contained a methylated RASSF1A gene (28), slightly lower than our findings. The same study found no RASSF1A methylation in normal breast. Lehman et al. (31) have found frequent methylation of RASSF1A in benign hyperplastic breast lesions and in intraductal hyperplasia but never in normal ductal epithelium. Similarly, our study found a significant frequency of RASSF1A methylation (34%) in benign breast lesions (intraductal papillomas), supporting the idea that RASSF1A methylation is an important event in abnormal breast epithelial proliferation.
Cyclin D2 methylation has been less studied in cancers. Little is known about cyclin D2’s involvement in breast cancer development, other than that transcription of cyclin D2 is down-regulated, both in sporadic and familial forms of cancer cases (32). Recent studies in gastric and pancreatic cancers showed that the methylation of cyclin D2 is responsible for its down-regulation in those two types of cancer (33, 34). The down-regulation is clearly due to its promoter methylation, which has been demonstrated in tumor cell lines and microdissected tumors. One recent study by Lehmann et al. (31) found cyclin D2 methylation, predominantly in DCIS (particularly high-grade) and invasive carcinomas. Another study by Evron et al. (24) found cyclin D2 methylation in 46% of breast carcinomas and associated this methylation with decreased cyclin D2 gene expression. Our work confirms that cyclin D2 methylation is a frequent event in breast carcinoma and is rare among intraductal papillomas. There was no significant difference in the frequency of cyclin D2 methylation among three histologic subtypes of invasive carcinoma, suggesting that these subtypes share a common carcinogenic mechanism involving cyclin D2.
Accurate diagnosis of malignancy in breast core biopsies and FNA biopsies is sometimes challenging and may require ancillary studies. We have successfully obtained gene methylation profiles from breast FNA samples, suggesting that application of MSP to clinical FNA samples is feasible. We also have found a high concordance of methylation profiles between breast FNAs and subsequent resection specimens, particularly for cyclin D2. Our study and others indicate that cyclin D2 methylation may provide a useful marker for malignancy in breast lesions because of its prevalence in carcinomas and infrequency in benign breast lesions. Future studies may identify other methylated genes that would complement a diagnostic panel of markers.
Finally, studies of gene methylation in breast carcinoma may also have therapeutic implications. Numerous drugs have been shown to alter gene methylation in vitro and in vivo, raising the possibility that reexpression of methylated genes in breast cancer may be useful. It has been shown that treatment of breast cancer cell lines with 5-Aza-2′-deoxycytidine (a demethylating agent) in conjunction with trichostatin (a histone deacetylase inhibitor) or all-trans-retinoic acid can induce reexpression of the estrogen receptor and RARβ2 (14, 35, 36). Future clinical trials may use MSP-based assays on FNA biopsy samples to identify patients who are eligible for gene reexpression therapy, based on their methylation profiles. Subsequent therapy with these agents may be monitored with repeated methylation profiles on FNA biopsies.
In short, methylation-specific PCR is a sensitive assay that is feasible for a diagnostic pathology laboratory; however, more work is clearly needed before methylation profiling becomes an adjunct to diagnostic pathology. The need to monitor new drugs that affect DNA methylation in tumors may also promote the development of clinical DNA methylation assays (26).
References
Baylin SB, Herman JG . Promoter hypermethylation—can this change alone ever designate true tumor suppressor gene function? J Natl Cancer Inst 2001; 93: 664–665.
Lapidus RG, Ferguson AT, Ottaviano YL, Parl FF, Smith HS, Weitzman SA, et al. Methylation of estrogen and progesterone receptor gene 5′ CpG islands correlates with lack of estrogen and progesterone receptor gene expression in breast tumors. Clin Cancer Res 1996; 2: 805–810.
Graff JR, Herman JG, Lapidus RG, Chopra H, Xu R, Jarrard DF, et al. E-cadherin expression is silenced by DNA hypermethylation in human breast and prostate carcinomas. Cancer Res 1995; 55: 5195–5199.
Evron E, Umbricht CB, Korz D, Raman V, Loeb DM, Niranjan B, et al. Loss of cyclin D2 expression in the majority of breast cancers is associated with promoter hypermethylation. Cancer Res 2001; 61: 2782–2787.
Bachman KE, Herman JG, Corn PG, Merlo A, Costello JF, Cavenee WK, et al. Methylation-associated silencing of the tissue inhibitor of metalloproteinase-3 gene suggest a suppressor role in kidney, brain, and other human cancers. Cancer Res 1999; 59: 798–802.
Esteller M, Corn PG, Urena JM, Gabrielson E, Baylin SB, Herman JG . Inactivation of glutathione S-transferase P1 gene by promoter hypermethylation in human neoplasia. Cancer Res 1998; 58: 4515–4518.
Herman JG, Merlo A, Mao L, Lapidus RG, Issa JP, Davidson NE, et al. Inactivation of the CDKN2/p16/MTS1 gene is frequently associated with aberrant DNA methylation in all common human cancers. Cancer Res 1995; 55: 4525–4530.
Dobrovic A, Simpfendorfer D . Methylation of the BRCA1 gene in sporadic breast cancer. Cancer Res 1997; 57: 3347–3350.
Ferguson AT, Evron E, Umbricht CB, Pandita TK, Chan TA, Hermeking H, et al. High frequency of hypermethylation at the 14-3-3 sigma locus leads to gene silencing in breast cancer. Proc Natl Acad Sci U S A 2000; 97: 6049–6054.
Widschwendter M, Berger J, Muller HM, Zeimet AG, Marth C . Epigenetic downregulation of the retinoic acid receptor-beta2 gene in breast cancer. J Mammary Gland Biol Neoplasia 2001; 6: 193–201.
Alvarez-Dolado M, Gonzalez-Sancho JM, Navarro-Yubero C, Garcia-Fernandez LF, Munoz A . Retinoic acid and 1,25-dihydroxyvitamin D3 inhibit tenascin-C expression in rat glioma C6 cells. J Neurosci Res 1999; 58: 293–300.
Chen GQ, Lin B, Dawson MI, Zhang XK . Nicotine modulates the effects of retinoids on growth inhibition and RAR beta expression in lung cancer cells. Int J Cancer 2002; 99: 171–178.
Swisshelm K, Ryan K, Lee X, Tsou HC, Peacocke M, Sager R . Down-regulation of retinoic acid receptor beta in mammary carcinoma cell lines and its up-regulation in senescing normal mammary epithelial. Cell Growth Differ 1994; 5: 2.
Widschwendter M, Berger J, Hermann M, Muller HM, Amberger A, Zeschnigk M, et al. Methylation and silencing of the retinoic acid receptor-beta2 gene in breast cancer. J Natl Cancer Inst 2000; 92: 826–832.
Virmani AK, Rathi A, Zochbauer-Muller S, Sacchi N, Fukuyama Y, Bryant D, et al. Promoter methylation and silencing of the retinoic acid receptor-beta gene in lung carcinomas. J Natl Cancer Inst 2000; 92: 1303–1307.
Yang Q, Mori I, Shan L, Nakamura M, Nakamura Y, Utsunomiya H, et al. Biallelic inactivation of retinoic acid receptor beta2 gene by epigenetic change in breast cancer. Am J Pathol 2001; 158: 299–303.
Sirchia SM, Ren M, Pili R, Sironi E, Somenzi G, Ghidoni R, et al. Endogenous reactivation of the RARbeta2 tumor suppressor gene epigenetically silenced in breast cancer. Cancer Res 2002; 62: 2455–2461.
Dreijerink K, Braga E, Kuzmin I, Geil L, Duh FM, Angeloni D, et al. The candidate tumor suppressor gene, RASSF1A, from human chromosome 3p21.3 is involved in kidney tumorigenesis. Proc Natl Acad Sci U S A 2001; 98: 7504–7509.
Dammann R, Li C, Yoon JH, Chin PL, Bates S, Pfeifer GP . Epigenetic inactivation of a RAS association domain family protein from the lung tumour suppressor locus 3p21.3. Nat Genet 2000; 25: 315–319.
Byun DS, Lee MG, Chae KS, Ryu BG, Chi SG . Frequent epigenetic inactivation of RASSF1A by aberrant promoter hypermethylation in human gastric adenocarcinoma. Cancer Res 2001; 61: 7034–7038.
Dammann R, Takahashi T, Pfeifer GP . The CpG island of the novel tumor suppressor gene RASSF1A is intensely methylated in primary small cell lung carcinomas. Oncogene 2001; 20: 3563–3567.
Lee MG, Kim HY, Byun DS, Lee SJ, Lee CH, Kim JI, et al. Frequent epigenetic inactivation of RASSF1A in human bladder carcinoma. Cancer Res 2001; 61: 6688–6692.
Takano Y, Kato Y, Masuda M, Ohshima Y, Okayasu I . Cyclin D2, but not cyclin D1, overexpression closely correlates with gastric cancer progression and prognosis. J Pathol 1999; 189: 194–200.
Evron E, Dooley WC, Umbricht CB, Rosenthal D, Sacchi N, Gabrielson E, et al. Detection of breast cancer cells in ductal lavage fluid by methylation-specific PCR. Lancet 2001; 357: 1335–1336.
Herman JG, Graff JR, Myohanen S, Nelkin BD, Baylin SB . Methylation-specific PCR. A novel PCR assay for methylation status of CpG islands. Proc Natl Acad Sci U S A 1996; 93: 9821–9826.
Pu RT, Clark DP . Detection of DNA methylation. Potential applications to diagnostic cytopathology. Acta Cytol 2003; 47: 247–252.
Burbee DG, Forgacs E, Zochbauer-Muller S, Shivakumar L, Fong K, Gao B, et al. Epigenetic inactivation of RASSF1A in lung and breast cancers and malignant phenotype suppression. J Natl Cancer Inst 2001; 93: 691–699.
Honorio S, Agathanggelou A, Schuermann M, Pankow W, Viacava P, Maher ER, et al. Detection of RASSF1A aberrant promoter hypermethylation in sputum from chronic smokers and ductal carcinoma in situ from breast cancer patients. Oncogene 2003; 22: 147–150.
Dammann R, Yang G, Pfeifer GP . Hypermethylation of the cpG island of Ras association domain family 1A (RASSF1A), a putative tumor suppressor gene from the 3p21.3 locus, occurs in a large percentage of human breast cancers. Cancer Res 2001; 61: 3105–3109.
Agathanggelou A, Honorio S, Macartney DP, Martinez A, Dallol A, Rader J, et al. Methylation associated inactivation of RASSF1A from region 3p21.3 in lung, breast and ovarian tumours. Oncogene 2001; 20: 1509–1518.
Lehmann U, Langer F, Feist H, Glockner S, Hasemeier B, Kreipe H . Quantitative assessment of promoter hypermethylation during breast cancer development. Am J Pathol 2002; 160: 605–612.
Fischer H, Chen J, Skoog L, Lindblom A . Cyclin D2 expression in familial and sporadic breast cancer. Oncol Rep 2002; 9: 1157–1161.
Matsubayashi H, Sato N, Fukushima N, Yeo CJ, Walter KM, Brune K, et al. Methylation of cyclin D2 is observed frequently in pancreatic cancer but is also an age-related phenomenon in gastrointestinal tissues. Clin Cancer Res 2003; 9: 1446–1452.
Yu J, Leung WK, Ebert MP, Leong RW, Tse PC, Chan MW, et al. Absence of cyclin D2 expression is associated with promoter hypermethylation in gastric cancer. Br J Cancer 2003; 88: 1560–1565.
Ferguson AT, Vertino PM, Spitzner JR, Baylin SB, Muller MT, Davidson NE . Role of estrogen receptor gene demethylation and DNA methyltransferase. DNA adduct formation in 5-aza-2′deoxycytidine-induced cytotoxicity in human breast cancer cells. J Biol Chem 1997; 272: 32260–32266.
Sirchia SM, Ferguson AT, Sironi E, Subramanyan S, Orlandi R, Sukumar S, et al. Evidence of epigenetic changes affecting the chromatin state of the retinoic acid receptor beta2 promoter in breast cancer cells. Oncogene 2000; 19: 1556–1563.
Acknowledgements
The authors thank Paul VanVelhuisen for help with the statistical analysis.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Pu, R., Laitala, L., Alli, P. et al. Methylation Profiling of Benign and Malignant Breast Lesions and Its Application to Cytopathology. Mod Pathol 16, 1095–1101 (2003). https://doi.org/10.1097/01.MP.0000095782.79895.E2
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1097/01.MP.0000095782.79895.E2
Keywords
This article is cited by
-
Clinical impact of PTEN methylation status as a prognostic marker for breast cancer
Journal of Genetic Engineering and Biotechnology (2021)
-
DNA methylation in ductal carcinoma in situof the breast
Breast Cancer Research (2013)
-
Epigenetics as a Therapeutic Target in Breast Cancer
Journal of Mammary Gland Biology and Neoplasia (2012)
-
High RASSF1A promoter methylation levels are predictive of poor prognosis in fine-needle aspirate washings of breast cancer lesions
Breast Cancer Research and Treatment (2011)
-
Promoter methylation and the detection of breast cancer
Cancer Causes & Control (2009)
