
Abstract
Tissue microarray technology facilitates rapid assessment of expression of molecular markers by enabling the simultaneous analysis of hundreds of tissue specimens. We have applied this technology to establish a microarray composed of cell pellets derived from 40 human lymphoma/leukemia-derived cell lines harboring a variety of molecular abnormalities. The application of cell line microarrays to the assessment of biologic marker evaluation was validated by studying the immunohistochemical expression of PTEN and phosphorylated AKT, two mediators of the phosphatidylinositol (PI)-3-kinase pathway. In addition to the high throughout analysis of protein expression in lymphoma/leukemia cells, this methodology also enables the evaluation of subcellular localization of protein expression. Cytoplasmic PTEN expression was observed in the majority of cell lines (87%), whereas a minor subset demonstrated nuclear expression. Phosphorylated AKT was also expressed predominantly within the cytoplasm in 65% of cell lines, whereas nuclear expression was seen in a minority. An inverse relationship between PTEN and phosphorylated AKT was observed in 63% of cell lines. No cell lines showed absence of PTEN expression, whereas 50% of cell lines showed low PTEN expression. Our data support the integrity of the PI-3-kinase-PTEN-AKT pathway in a majority of cell lines derived from hematologic malignancies and clearly demonstrates the utility of microarray technology in the in situ assessment of expression of molecular markers in tumor-derived cell lines.
Similar content being viewed by others

INTRODUCTION
Tissue microarray technology permits simultaneous analysis of hundreds of tissue specimens (1, 2, 3). Tissue microarrays are powerful for validation, prioritization, and extension of findings obtained from genomic surveys, such as cDNA microarrays (4) at the DNA, RNA, or protein level (5). They have been successfully used for high-throughput molecular profiling of a variety of cancers including breast, prostate, bladder, and lymphoma (6, 7, 8, 9). Technical considerations regarding the establishment of tissue microarrays have been amply reported; however, application of such technology to the study of pure tumor cell pellets derived from cells grown in continuous culture has not been adequately addressed in the literature. In this study, we modified the tissue microarray approach to establish a cell line microarray composed of cell pellets obtained from human tumor–derived cell lines. This method would faciliate the in situ analysis of pure tumor populations for expression of biomarkers at the DNA, RNA, and protein level.
In this study, we have applied the tissue microarray technology to analyze the in situ expression of two mediators of the phosphatidylinositol (PI)-3-kinase pathway, PTEN and AKT, in microarrays composed of 40 cell lines derived from hematopoietic neoplasms. The tumor suppressor gene, PTEN, is deregulated in a variety of cancers (10). It encodes a dual-specificity phosphatase with homology to the cytoskeletal proteins tensin and auxilin (11). It is unique in that it has phosphatase activity against protein tyrosine kinases as well as phosphatidylinositol-3, 4, 5-trisphosphates (11). The protein tyrosine and lipid phosphatase activity of PTEN places it as a key regulator of the effects of signaling from receptor tyrosine kinases via phosphatidylinositol-3-kinase to PKB/AKT and its downstream targets such as BAD, thus influencing cell proliferation, survival, differentiation, motility, and invasiveness (12).
The cellular mechanism of PTEN function and its role in the molecular pathogenesis of malignant lymphoma are unknown. There is a high incidence of hematopoietic tumors and lymphoid hyperplasia in PTEN-heterozygous mice (13), suggesting the role of PTEN in regulating hematopoietic cell proliferation, cell death, and malignant transformation. The PI3-K/AKT pathway is activated in many lymphomas; however, comprehensive analysis of PTEN and AKT expression at the protein level has not been performed in these cancers. Many studies have reported a relatively low incidence of loss of heterozygosity and point mutations of PTEN in hematopoietic neoplasms (14). It is noteworthy that most studies of PTEN in lymphomas have searched exclusively for deletions or mutations of the gene rather than for dysregulation of mRNA or protein expression. As a key inhibitor of the PI-3-kinase, an inverse relationship between PTEN and phosphorylated AKT has been demonstrated (15) in a variety of cancers by Western blot analysis and tissue immunohistochemistry (16, 17). A major limitation of Western blot analysis is the loss of subcellular expression information. The expression of clinically relevant molecular biomarkers at the microanatomical level is of critical importance. By analysis of PTEN and phosphorylated AKT expression, we demonstrate the integrity of the PI-3-kinase-PTEN-AKT pathway in a majority of cell lines derived from hematologic malignancies. This is the first report of a microarray approach to study a comprehensive panel of tumor-derived cell lines for analysis of molecular marker expression.
MATERIALS AND METHODS
Microarrays of cores prepared from cell blocks were obtained using cell lines derived from 40 hematologic malignancies harboring a variety of molecular abnormalities: T- and B-acute lymphoblastic lymphoma, t(14;18)–positive large B-cell lymphoma, t(11;14)–positive mantle cell lymphoma, t(9;22)–positive chronic myeloid leukemia, t(15;17)–positive acute myeloid leukemia, cutaneous T-cell lymphoma, t(2;5) positive anaplastic large cell lymphoma, t(8;14)–positive Burkitt lymphoma, Hodgkin's lymphoma, and t(14;18)–negative large B-cell lymphoma). Table 1 summarizes the molecular abnormalities associated with the hematopoietic neoplasms. Cell lines were obtained from the American Tissue Culture Collection (Rockville, MD) or from the laboratory of Dr. Hans Messner, University of Toronto. Cells were grown in RPMI 1640 (Nova-Tech, Inc.) supplemented with 1.6 g/L sodium bicarbonate, 4.5 g/L glucose, 9.5 g/L HEPES, 0.11g/L sodium pyruvate, and 10–20% heat-inactivated fetal bovine serum. Each cell line was grown to approximately the concentration of 20 to 25 million cells in 50 mL of media. Cell viability was determined by trypan blue dye exclusion (Sigma), and the desired standard of cell viability was 90% for each cell line.
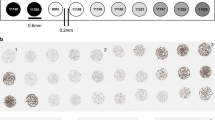
For paraffin-block preparation, the cells were pelleted using low-speed centrifugation at 1500 rpm for 10 minutes. The supernatant was decanted, and normal plasma and thromboplastin were added in equal amounts (six drops) and allowed to congeal for 5 minutes. The clot was placed into tissue paper, folded into a biopsy cassette, and fixed in 10% neutral buffered formalin. The cassette was then submitted for routine histological processing. Construction of the tissue microarray block was achieved by making 2-mm holes in a recipient tissue microarray block, acquiring a 2-mm cylindrical core specimen from cell blocks of all 40 hematopoietic cell lines and depositing them into the recipient tissue microarray block. These cores were first organized into groups of related malignancies and then arrayed into a recipient tissue microarray block. Figure 1A demonstrates the array of cell blocks after staining with hematoxylin-eosin. Five replicate tissue microarray blocks were created with the position of the specimens at identical coordinates in each block. Alternatively, several replicate cores of one cell block can be placed in one recipient block.

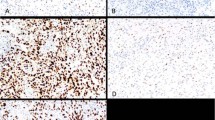
A, an array of hematoxylin-eosin stained cell blocks prepared from 40 cell lines derived from hematopoietic neoplasms outlined in Table 1. B, a higher magnification of a representative cell block core. C, a cell line (SUDHL-9) expressing low levels of PTEN. D, a cell line (SUDHL-2) expressing high levels of cytoplasmic PTEN. E, illustrates a cell line (HPB) with low expression of phosphorylated AKT. F, high levels of phosphorylated AKT protein is seen in a cell line (BC-3). There is a submembranous accentuation in the cells showing positive reactivity for phosphorylated AKT (E), whereas a strong Golgi-associated pattern is seen with PTEN (D).
For immunohistochemical studies, 5-μm-thick serial sections were mounted on glass slides coated with 2% aminopropyltrioxysilane (APES; Sigma Corp., St. Louis, MO) in acetone. Sections were dewaxed in xylene and rehydrated in graded ethanols. Endogenous peroxidase activity was blocked by immersion in 0.3% methanolic peroxide for 15 minutes. The immunoreactivity of the target antigens was enhanced by microwave incubation of the tissue sections for 15 minutes in 0.1 m citrate buffer. Expression of PTEN and phosphorylated AKT was determined by immunohistochemical analysis using a rabbit polyclonal antibody for PTEN (Zymed, San Fransisco, CA) at a 1:50 dilution and a polyclonal antibody for Ser473-phosphorylated AKT (Cell Signaling Technology, Beverly, MA) at a dilution of 1:25. The sections were then incubated with a biotinylated peroxidase agent, a 3,3′-diaminobenzidine basic detection kit (Ventana Medical Systems Inc., Tucson, AZ). Antigen–antibody reactions were visualized using diaminobenzidine as the chromogen. Normal mouse serum containing mixed immunoglobulins at a concentration approximating that of the primary antibody was used as a negative control. Sections were counterstained with hematoxylin. Prostate epithelium was used as an external positive control for both antibodies and endothelial cells represented internal controls.
A semi-quantitative assessment of intensity was performed by two evaluators (MSL and RA) and divided into two grades; 1+ (low expression defined as less than expression level seen in prostate epithelium) and 2+ (high expression defined as equal to or greater than expression level seen in prostate epithelium). Subcellular localization of protein expression was also evaluated.
RESULTS
Cell Line Microarray
Table 1 summarizes the key molecular abnormalities associated with the hematopoietic cell lines used in the creation of the microarray. Cell pellets were resuspended in normal plasma and thromboplastin to facilitate cell–cell integrity, enabling easy cutting of microarray blocks. Limiting the use of cell cultures demonstrating >90% viability by trypan blue dye exclusion ensured adequate numbers of viable cells for subsequent morphologic analysis. Starting with 20–25 million cells in 50 mL of media (0.4–0.5 million cells per milliliter), we obtained approximately 500 cells per core. Under these conditions, optimal cell morphology was obtained without overlapping of cellular detail due to crowding. The cell lines were organized into relevant groups of neoplasms, such as T- and B-acute lymphoblastic leukemias, t(14;18)–positive diffuse large B-cell lymphomas, t(11;14)–positive mantle cell lymphoma, t(9;22)–positive chronic myeloid leukemia, t(15;17)–positive acute myeloid leukemia, cutaneous T-cell lymphoma, t(2;5)–positive anaplastic large cell lymphoma, t(8;14)–positive Burkitt lymphoma, Hodgkin lymphoma, and t(14;18)–negative large B-cell lymphoma, which facilitates the correlation with biomarker expression.
Figure 1A demonstrates a hematoxylin-eosin–stained array of 40 2.0-mm-diameter cell blocks prepared from a wide spectrum of hematologic malignancies. With a starting population of approximately 20–25 million cells, we were able to construct five replicate microarray blocks. Up to 30 consecutive sections could be cut from each of the blocks, resulting in a total of 150 sections. Microscopic analysis of the cell block shown in Figure 1B clearly demonstrated the presence of >500 cells in >90% of the arrays. With the analysis of two cell arrays, we were able to analyze adequate numbers of cells (>500 cells per core) in 40/40 (100%) of the cell blocks.
Immunohistochemical Analysis
Routine immunohistochemical analysis for PTEN and phosphorylated AKT was performed using a similar protocol for paraffin-embedded formalin-fixed tissue blocks. Variable levels of PTEN protein were detected in the cell lines. We compared the intensity of PTEN expression to that seen in external control prostate tissue samples where prostate epithelium exhibited high PTEN (2+) expression. The data are summarized in Table 1. PTEN expression was observed in the cytoplasm of the majority of cell lines (87%), whereas a minor subset demonstrated nuclear expression. Thirty-two percent of cell lines showed low (1+) PTEN expression, whereas the other 68% showed high (2+) PTEN expression. No cell lines showed complete absence of PTEN expression, consistent with previous reports of low incidence of PTEN gene alterations in leukemias and lymphomas (18). In a subset of cell lines, a characteristic Golgi-staining pattern was observed (Figure 1D).
Phosphorylated AKT was also expressed predominantly within the cytoplasm in 65% of cell lines, whereas nuclear expression was seen in a minority. The majority of cell lines, 28/40 (70%), demonstrated low levels (1+) of phosphorylated AKT, whereas 12 cell lines (30%) showed high (2+) phosphorylated AKT. An inverse relationship between PTEN and phosphorylated AKT was observed in 63% of cell lines. Eight cell lines demonstrated low levels of both PTEN and phosphorylated AKT, whereas seven cell lines demonstrated high levels of both proteins. Figure 1C demonstrates a cell line with low PTEN expression, whereas Figure 1D demonstrates high levels of cytoplasmic expression of PTEN, with a Golgi-staining pattern. Figure 1E is an example of a cell line with low levels of phosphorylated AKT. Figure 1F demonstrates a cell line with high levels of phosphorylated AKT highlighting its submembranous localization. Detection of PTEN and phosphorylated AKT protein expression within the nucleus (Fig. 1E, inset) of a subset of cell lines supports the existing literature that abnormal nucleocytoplasmic shuttling of key signaling molecules may play a role in the development of malignant neoplasms (16).
DISCUSSION
We have applied the use of tissue microarray technology to analyze the in situ expression of two mediators of the phosphatidylinositol (PI)-3-kinase pathway, PTEN and AKT, in 40 hematopoietic cell lines. To date, human tissue microarrays are most commonly constructed from archival paraffin tissue blocks. Arrays composed of frozen tumor tissues have also been demonstrated to enhance the analysis of RNA by in situ hybridization, FISH, and immunohistochemistry (5). Microarrays of tissue samples, however, are not amenable to studying changes in gene or protein expression in response to growth factors, kinase inhibitors, or chemotherapeutic agents. The availability of a microarray composed of tumor-derived cell lines would be highly useful in initial validation of cDNA microarray and proteomic data. Although we carried out our studies with hematopoietic cells that grow in suspension, adherent tumor cells such as carcinomas and sarcomas can also be used after trypsin disaggregation. Control populations of cells such as quiescent and activated peripheral blood T and B lymphocytes may also be included in the microarray block. Additionally, this approach would allow the study of cells that have been exposed to the effects of varying microenvironmental stimuli, such as co-culture of tumor cells with fibroblasts and endothelial cells. The determination of intracellular localization of gene products would also provide critical information regarding their biologic function. The delineation of signaling pathways and gene networks that are increasingly being implicated in tumorigenesis through genomic and proteomic studies (19, 20) highlights the need for multiplex biomarker analysis. Cell arrays created from tumor-derived cell lines can provide starting material to analyze DNA, RNA, and proteins, as well as phospho-proteins.
Although 20–25 million cells were used for preparation of each cell block in our study, microscopic analysis of the cell blocks clearly demonstrated that this number was more than sufficient for routine immunohistochemical analysis. We and others (5) have demonstrated that 0.6-mm-diameter cell blocks would be equally sufficient for routine RNA, DNA, and protein analysis. Furthermore, because the mode of fixation can be optimized to the specific procedure to be used for analysis, there is an added level of flexibility in the nature of the study, such as RNA in situ hybridization, FISH, and immunohistochemistry.
By analysis of PTEN and phosphorylated AKT expression, we demonstrate the integrity of the PI-3-kinase-PTEN-AKT pathway in a majority of cell lines derived from hematologic malignancies. We were able to confirm in a relatively rapid manner that PTEN deletions or mutation resulting in absence of PTEN expression occurs rarely in hematopoietic neoplasms (18). This is the first report to demonstrate in situ phosphorylated AKT expression in a comprehensive group of hematologic malignancies. Detection of PTEN and phosphorylated AKT protein expression within the nucleus of a subset of cell lines supports the existing literature that abnormal nucleocytoplasmic shuttling of key signaling molecules may play a role in the development of malignant neoplasms.
In summary, tumor-derived cell lines provide an excellent target material for the study of DNA, RNA, and proteins and clearly demonstrate the ability of tissue microarray technology in linking protein expression with previously identified molecular abnormalities. This approach will allow rapid and high-throughput assessment of changes in multiple gene expression in response to therapeutic agents directed against specific molecular defects.
References
Bubendorf L, Nocito A, Moch H, Sauter G . Tissue microarray (TMA) technology: miniaturized pathology archives for high-throughput in situ studies. J Pathol 2001; 195: 72–79.
Hoos A, Cordon-Cardo C . Tissue microarray profiling of cancer specimens and cell lines: opportunities and limitations. Lab Invest 2001; 81: 1331–1338.
Kallioniemi OP, Wagner U, Kononen J, Sauter G . Tissue microarray technology for high-throughput molecular profiling of cancer. Hum Mol Genet 2001; 10: 657–662.
Barlund M, Forozan F, Kononen J, Bubendorf L, Chen Y, Bittner ML, et al. Detecting activation of ribosomal protein S6 kinase by complementary DNA and tissue microarray analysis. J Natl Cancer Inst 2000; 92: 1252–1259.
Fejzo MS, Slamon DJ . Frozen tumor tissue microarray technology for analysis of tumor RNA, DNA, and proteins. Am J Pathol 2001; 159: 1645–1650.
Camp RL, Charette LA, Rimm DL . Validation of tissue microarray technology in breast carcinoma. Lab Invest 2000; 80: 1943–1949.
Manley S, Mucci NR, De Marzo AM, Rubin MA . Relational database structure to manage high-density tissue microarray data and images for pathology studies focusing on clinical outcome: the prostate specialized program of research excellence model. Am J Pathol 2001; 159: 837–843.
Richter J, Wagner U, Kononen J, Fijan A, Bruderer J, Schmid U, et al. High-throughput tissue microarray analysis of cyclin E gene amplification and overexpression in urinary bladder cancer. Am J Pathol 2000; 157: 787–794.
Natkunam Y, Warnke RA, Montgomery K, Falini B, van De Rijn M . Analysis of MUM1/IRF4 protein expression using tissue microarrays and immunohistochemistry. Mod Pathol 2001; 14: 686–694.
Li J, Yen C, Liaw D, Podsypanina K, Bose S, Wang SI, et al. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science 1997; 275: 1943–1947.
Maehama T, Dixon JE . The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-trisphosphate. J Biol Chem 1998; 273: 13375–13378.
Furnari FB, Huang HJ, Cavenee WK . The phosphoinositol phosphatase activity of PTEN mediates a serum-sensitive G1 growth arrest in glioma cells. Cancer Res 1998; 58: 5002–5008.
Tamura M, Gu J, Matsumoto K, Aota S, Parsons R, Yamada KM . Inhibition of cell migration, spreading, and focal adhesions by tumor suppressor PTEN. Science 1998; 280: 1614–1617.
Sakai A, Thieblemont C, Wellmann A, Jaffe ES, Raffeld M . PTEN gene alterations in lymphoid neoplasms. Blood 1998; 92: 3410–3415.
Hyun T, Yam A, Pece S, Xie X, Zhang J, Miki T, et al. Loss of PTEN expression leading to high Akt activation in human multiple myelomas. Blood 2000; 96: 3560–3568.
Gimm O, Perren A, Weng LP, Marsh DJ, Yeh JJ, Ziebold U, et al. Differential nuclear and cytoplasmic expression of PTEN in normal thyroid tissue, and benign and malignant epithelial thyroid tumors. Am J Pathol 2000; 156: 1693–1700.
Kanamori Y, Kigawa J, Itamochi H, Shimada M, Takahashi M, Kamazawa S, et al. Correlation between loss of PTEN expression and Akt phosphorylation in endometrial carcinoma. Clin Cancer Res 2001; 7: 892–895.
Sakai A, Thieblemont C, Wellmann A, Jaffe ES, Raffeld M . PTEN gene alterations in lymphoid neoplasms. Blood 1998; 92: 3410–3415.
Hanash SM . Global profiling of gene expression in cancer using genomics and proteomics. Curr Opin Mol Ther 2001; 3: 538–545.
Simpson RJ, Dorow DS . Cancer proteomics: from signaling networks to tumor markers. Trends Biotechnol 2001; 19: S40–S48.
Author information
Authors and Affiliations
Corresponding author
Additional information
Support for this article was provided in part by grants from the National Leukemia Foundation to MSL and by a grant (CA 83984–01) from the National Institutes of Health to KSJ-EJ.
Rights and permissions
About this article
Cite this article
Abbott, R., Tripp, S., Perkins, S. et al. Analysis of the PI-3-Kinase-PTEN-AKT Pathway in Human Lymphoma and Leukemia Using a Cell Line Microarray. Mod Pathol 16, 607–612 (2003). https://doi.org/10.1097/01.MP.0000067423.83712.74
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1097/01.MP.0000067423.83712.74
