
Abstract
Epstein-Barr virus (EBV) is associated with a substantial number of gastric adenocarcinomas worldwide, as confirmed by EBER1/2-RNA in situ hybridization (RISH). In the present study, we developed a rapid and sensitive PCR-based prescreening method for the detection of EBV in gastric carcinomas to reduce the amount of laborious EBER1/2-RISH assays to be performed. The method was evaluated by testing gastric adenocarcinomas (n = 242) using both BamHI W PCR-enzyme immunoassay (EIA) and EBER1/2-RISH, in combination with appropriate DNA and RNA quality controls. Seventy-four percent of the paraffin-embedded gastric adenocarcinomas had good DNA quality as shown by β-globin polymerase chain reaction (PCR) after proteinase K and boiling pretreatment, whereas after DNA purification this was increased to 90%. Thirty-two percent of all cases were EBV-DNA positive after PCR-EIA, whereas 10% of these gastric cancers contained EBV transcripts in the neoplastic cells as confirmed by EBER1/2-RISH. Interestingly, only samples with high optical density (OD) 405/630 values in PCR-EIA, equivalent to the maximum reading of the assay as determined by the positive control, contained EBV-positive tumor cells in the EBER1/2-RISH. In contrast, the weak positive samples, as determined by low OD readings in the PCR-EIA were EBER1/2-RISH negative. In conclusion, high OD values in EBV PCR-EIA are very valuable to prescreen EBV-carrying gastric carcinomas as confirmed by EBER1/2-RISH. Only these samples and those with poor DNA quality will require testing in the EBER1/2-RISH, thereby reducing the amount of laborious RISH assays with 85%.
Similar content being viewed by others


INTRODUCTION
In recent years, the human pathogenic Epstein-Barr virus (EBV) has been detected in a subset of gastric carcinomas. This ubiquitous human γ-herpes virus is known to cause several benign and malignant diseases of both lymphoid and epithelial origin in humans (1). EBV can be detected in 75 to 100% of the relatively rare lymphoepithelioma-like gastric carcinomas (2, 3, 4) and in approximately 27 to 35% of gastric stump carcinomas (5, 6). In addition, EBV is detected in approximately 10% (range, 2–16%) of gastric adenocarcinomas n.o.s. (not otherwise specified) using EBER1/2-RNA in situ hybridization (EBER1/2-RISH; 7, 8, 9). It has been shown that EBV is clonally present in all tumor cells (10, 11) and transcriptionally active (12, 13). The pathogenic role of EBV in these tumors remains to be established. Therefore, large epidemiological and clinicopathological studies are required.
At present, the nonradioactive EBER1/2-RISH is considered the most reliable technique, allowing the identification of EBV-bearing tumor cells in context of the morphology of tissue sections (14, 15, 16). The two EBV-encoded RNA transcripts 1 and 2 (EBER1/2) do not encode proteins, but they might be important for oncogenesis and resistance to programmed cell death (17). These EBER1/2-transcripts are a valuable screening tool because they are abundantly expressed in all latency patterns displayed in tumors (18, 19). However, EBER1/2-RISH is a laborious technique, in which only small amounts of samples can be tested per assay. In addition, its evaluation requires trained personnel.
In recent years, DNA-PCR has been described as a valuable method for the detection of EBV in malignancies (20, 21). However, detection does not take place at the single-cell level. It is therefore not possible to determine the source of the detected EBV-DNA to exclude amplification of EBV from other sources than the tumor cells.
The aim of this study was to develop a rapid and reliable PCR-based hybridization assay facilitating prescreening of clinical samples. The sensitive semiquantitative PCR for the BamHI W repeat was used in combination with a phosphatase-based enzyme immunoassay. In addition, all cases were also tested by EBER1/2-RISH. We show that PCR-EIA can be used as a reliable prescreening method and that the presence of EBV in the tumor cells only has to be confirmed for strong EBV-DNA positive cases or for cases with poor DNA quality.
MATERIALS AND METHODS
Clinical Material
Two hundred forty-two cases of formalin-fixed and paraffin-embedded resection material or biopsies of gastric adenocarcinomas were tested of which 120 were obtained from the Department of Pathology of the Vrije Universiteit medical center and 122 gastric adenocarcinomas from 18 hospitals in the Netherlands, collected during the large Dutch D1D2 Gastric Cancer study conducted at the Leiden University Medical Center (22). Tumor blocks were selected based on pathology reports.
Pretreatment and DNA Isolation
Sandwich sections of 5 μm (outer sections) were stained with hematoxylin and eosin to confirm the presence of tumor in the tissue. For PCR purposes, a total amount of sections (5 μm), in between sections, representing 1-cm2 tumor were used. The tissue sections were digested overnight at 37° C using 100 μg/mL proteinase K and 0.45% Tween-20 in 10 mm Tris–HCl (pH 7.5), followed by inactivation of proteinase K and lysis of the cells by boiling the samples for 10 minutes (23). Samples were centrifuged for 5 minutes at maximum speed in a tabletop centrifuge and placed on ice. Ten microliters of this suspension was used in the PCR.
An additional DNA isolation procedure was performed on 200-μL proteinase K mixture of those samples that were initially negative in the β-globin PCR to remove potentially PCR-inhibiting factors using the High Pure PCR Template Purification (HPPTP) kit (Roche, Indianapolis, IN) according to the manufacturer's protocol. Ten microliters of the 200-μL elute was used for PCR.
β-Globin PCR
Samples were analyzed for proper DNA quality for amplification by the human β-globin PCR using the BGPCO3.1–BGPCO5 primer set, as described elsewhere (24). The 209-bp PCR products were visualized by gel electrophoresis on a 1.5% agarose gel and evaluated compared with the positive control (1 × 104 JY cells per reaction).
Epstein-Barr Virus PCR
For the detection of EBV, the DNA isolate was subjected to EBV14/20 BamHI W repeat PCR as described previously (25), except that the EBV20 primer was biotinylated at the 5′ end during oligonucleotide synthesis (bioEBV20: 5′ bio-gtgaagtcacaaacaagcc-3′; EBV 14: 5′ ctctggtagtgatttggacc-3′). Distilled water was included as a negative control, whereas a dilution range of JY cells (100 BamHI W targets/cell) was included as a control for the sensitivity of the assay.
EBV PCR products of 240 bp were hybridized in the newly developed BamHI W enzyme immunoassay, according to a procedure previously described for the detection of human papilloma virus DNA (26). Five-microliter biotinylated BamHI W-PCR product was coated onto a 96-well streptavidin-coated microtiter plate and 50-μL of washing buffer, containing 1× SSC and 0.5% Tween-20, was added to each well. The samples were incubated for 1 hour at 37° C. The microtiter plate was washed four times (350 μL; 30 seconds) with washing buffer using an automated plate washer. The samples were denaturated for 15 minutes at room temperature using 0.2 m NaOH, washed four times, and hybridized with the digoxigenin-labeled pEBV probe (5′ Dig-aatctgacactttagagctctggaggactt-3′; Eurogentec, Liege, Belgium), at a concentration of 10 pmol/mL of washing buffer, for 60 minutes at 37° C. After washing the plates four times, the samples were incubated for another hour with the alkaline phosphatase–labeled anti-dig Fab fragments. Samples were washed five times and visualized using the pNPP substrate (Sigma, St. Louis, MO). Optical density (OD) 405/630 values were measured after 1 hour and overnight substrate incubation. Samples were evaluated and compared with the appropriate controls. The cut off value was defined as three times the average negative control (26). The OD of the samples was compared with the OD of the JY dilution range. In our setting, maximal OD reading was obtained using 102 JY cells (103 EBV copies) after overnight substrate incubation.
EBER1/2 and β-Actin RNA In Situ Hybridization
RNA in situ hybridization was performed for the EBER1/2-transcripts and the β-actin target as described previously (27). Briefly, 5-μm sections of paraffin-fixed tissue were cut and sections for β-actin-RISH were pretreated with proteinase K (10 μg/mL). Sections were hybridized overnight at 55° C in a mix containing 50% formamide (Merck, Whitehouse Station, NJ), 10% dextran sulfate (Eppendorf 5 prime, Boulder, CO), 0.5× SSC (0.2× SSC for β-actin-RISH) and digoxigenin-labeled EBER1 and EBER2 probes diluted 1:100 (1:50 for β-actin-RISH). Excess probe was washed away by rinsing the slides three times for 15 minutes each with 0.5× SSC. Bound probe was detected using mouse anti-digoxigenin (Sigma), biotinylated rabbit anti-mouse (DAKO, Glostrup, Denmark), followed by streptavidin–biotin complex (SABC, DAKO) and catalyzed reporter deposition signal enhancement. Aminoethylcarbazol was used for visualization of EBER1/2-RISH slides, whereas β-actin-RISH slides were visualized using diaminobenzidine nickel.
EBER1/2-RISH and Immunohistochemistry: Double Staining
Double staining for EBER1/2-transcripts and keratin was performed as described elsewhere (28). Briefly, EBER1/2-RISH was performed and the slides were visualized using diaminobenzidine, followed by staining with the Moab AE 1/3 for keratin (Boehringer Mannheim, Mannheim, Germany) and visualization by Fast Blue BB Base substrate (Sigma, St. Louis, MO).
Statistical Analyses
For statistical testing, the two-tailed Fisher exact test was used. A P value of <0.05 was considered statistically significant.
RESULTS
Polymerase Chain Reaction
All 242 samples had tumor cells in sandwich HE sections and were tested by β-globin and EBV PCR. Results are shown in Table 1. As determined by β-globin PCR, 74% (178/242) of the samples had good DNA quality for PCR amplification after treatment with proteinase K and boiling. EBV DNA prevalence in these samples, regardless of DNA quality, was 26.4% (64/242).
Quantitative analysis of the OD readings of PCR-EIA EBV-positive group allowed the division of these cases into two subgroups: weak positive (+) and strong positive (++). Strong positive cases were defined as those that reached a maximum OD reading after overnight substrate incubation. This OD value was identical to the OD of 102 JY cells (103 EBV copies). In our setting, the OD of this positive control ranged on average from 2.300–2.500. In contrast, weak-positive cases reached OD readings higher than the cutoff level as determined by three times the OD reading of the negative control, but lower than the maximal OD reading. These OD values, except for one case, never exceeded 2.000 after overnight substrate incubation.
After DNA purification of samples with poor DNA quality for PCR amplification, the β-globin PCR positivity was increased to 90% (218/242) and overall positivity in EBV-DNA PCR to 31.8% (77/242), including 9 weak EBV-positive cases and 4 strong EBV-positive cases. In addition, a quantitative increase in EBV PCR-EIA positivity was observed, as determined by higher OD readings in EBV-positive cases with poor DNA quality previously, including one weak EBV-positive case showing a strong positive signal.
EBER1/2-RNA In Situ Hybridization
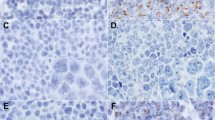
All 242 cases were tested for the presence of EBV in the tumor cells by EBER1/2-RISH (Table 2). Ten percent (25/242) of these tumors were EBV positive. They were characterized by nuclear staining of all tumor cells with the EBER1 and EBER2 anti-sense probes (Fig. 1A). Specificity of the hybridization was confirmed for each case using EBER1 and EBER2 sense probes as a negative control (Fig. 1B). In two instances, positivity in the sense control was observed, which was most likely caused by aspecific probe hybridization, and therefore these cases were considered EBV negative. Indeed, this was confirmed by the negative EBV-PCR status of these cases. In addition, infiltrating lymphocytes were occasionally positive for EBV transcripts. These cases were not considered as EBV-carrying gastric carcinomas. In a subset of these cases the lymphoid character of these cells was confirmed by EBER1/2-RISH and keratin IH double staining (Fig. 2).

Representative RNA in situ hybridization (RISH) of Epstein-Barr virus–carrying gastric carcinoma. A, EBER1/2-RISH antisense: showing red nuclear staining in all tumor cells. B, EBER1/2-RISH sense control. C, β-actin-RISH antisense; cytoplasmic staining (black) of all cells. D, β-actin-RISH sense control.

Double staining for EBER1/2-RISH (brown) and keratin-IH (blue). A, section of the slide showing EBER1/2-RISH–negative tumor glands (magnification, 200×). B, magnification of a section showing a EBER1/2-RISH positive infiltrating lymphocyte (magnification, 400×).
β-Actin-RISH was included as RNA quality control for a subset of 111 cases. Data are shown in Table 3. Eighty-eight percent (98/111) of the tumor tissues tested showed good RNA quality enabling the detection of potentially present EBER1/2-transcripts, as determined by cytoplasmic staining of the whole tissue section (Fig. 1C), whereas the sense control was negative (Fig. 1D). Five percent (6/111) of the cases showed RNA degradation, which would make proper detection of EBV transcripts impossible. In 7% of the cases on which β-actin-RISH was performed, interpretation was not possible because of background in the sense control.
Correlation PCR Versus RISH
A very interesting correlation was observed between the OD readings in the PCR-EIA and the EBER1/2-RISH data, as shown in Table 2. Strong positive EBV-DNA signals correlated in 23 out of 24 cases with EBV presence in the tumor cells, whereas EBV PCR-EIA negative results of samples with proper DNA quality as determined by β-globin PCR, correlated in 100% with absence of virus the tumor. In addition, in all but one of the cases with weak positive hybridization signals, EBV was not detected by EBER1/2-RISH. β-Actin-RISH was performed on all weak positive cases and showed good RNA quality, therefore indicating true absence of EBV in these tumor cells. Statistical analysis showed a significant correlation (P<.0001) between the strength of the EIA positivity (weak versus strong) and the EBER1/2-RISH results as determined by the two-tailed Fisher's exact test.
DISCUSSION
In the present study, we have shown that the BamHI W PCR-EIA in combination with a DNA quality control is a very valuable tool in rapidly and reliably identifying EBV-carrying gastric carcinomas, as confirmed by EBER1/2-RISH. Thus, a screening method for the detection of EBV has been established, which is significantly less laborious and time consuming than the conventionally performed EBER1/2-RISH.
We observed that strong hybridization signals in the PCR-EIA were restricted to EBER1/2-RISH positive gastric carcinomas, when testing all cases by both PCR and RISH. Based on this observation, we developed and evaluated our rapid and reliable PCR-based prescreening protocol in which only strong EBV-DNA positive cases and those with poor DNA quality require testing by EBER1/2-RISH (Fig. 3). Meanwhile, we have applied this screening method to an additional 82 randomly selected cases, and observed an overall EBV positivity of 7.7% in a total of 324 cases from 19 different Dutch hospitals (data not shown). This is in agreement with the current literature concerning the prevalence of EBV-carrying gastric adenocarcinomas worldwide (7, 9, 29, 30).

Approach for screening of gastric adenocarcinomas.
Using the combination of PCR-EIA and EBER1/2-RISH, we were able to determine that the weak-positive signals in PCR-EIA correlated with sporadically infiltrating EBER1/2-RISH positive lymphocytes. Double staining for EBER1/2-transcripts and keratin confirmed the lymphoid origin of these cells (Fig. 2). However, consecutive sections were used for PCR and RISH, explaining discrepancies observed in the separate assays. In contrast to EBER1/2-RISH–negative gastric carcinomas containing randomly dispersed tumor infiltrating EBER1/2-RISH–positive lymphocytes, EBV-carrying gastric carcinomas are characterized by the presence of EBV in all tumor cells. This fact was reflected by the high OD readings after PCR-EIA and allowed us to use these high OD readings as screening method.
One prerequisite for the application of PCR-based screening methods is sufficient DNA of proper quality for PCR amplification. Therefore, all samples were tested by β-globin PCR and, if necessary, DNA purification was performed. After DNA purification, 90% of the cases tested did show appropriate DNA quality for PCR amplification. PCR-inhibiting factors or DNA degradation were probably caused by fixation and processing of the tissue, which might differ in the different pathology departments. This indicates that this method is suitable even for different tissue fixation techniques. Additional methods to increase the DNA quality for PCR as reported, for instance, by Satoh et al. (31) by pretreatment of gastric DNA isolates with restriction enzymes or bovine serum albumin, have not been used in our study.
We evaluated all cases, including the EBV-DNA negative cases by EBER1/2-RISH, thereby confirming the sensitivity of the PCR assay. All EBV PCR-negative cases with proper DNA quality were EBER1/2-RISH negative. In addition, in one of the β-globin–negative cases, an additional EBER1/2-RISH–positive gastric carcinoma was found. This case could be detected because of the inclusion of a DNA quality control in the protocol, thereby supporting the value of checking DNA quality. In addition, weak EBV-positive cases did not contain EBV transcripts in the tumor cells, except for one case. EBV positivity in the PCR-EIA was followed in all cases by EBER1/2-RISH to confirm the presence of EBV in the tumor cells.
To exclude the possibility that EBER1/2-RISH–negative tumors were a consequence of poor RNA-quality of formalin-fixed and paraffin-embedded tissue, a β-actin-RISH was carried out. This procedure allowed a thorough evaluation of the EBER1/2-RISH results and has proven to be a suitable tool to determine false negative results. However, this method did not increase the specificity of the assay but did increase the numbers of control assays to be performed. Therefore, β-actin-RISH will not be included in our standard screening protocol. In contrast, the EBER1/2-sense control was shown to be informative in two instances and will therefore remain in the strategy.
Previous studies have evaluated the use of PCR and ISH for other EBV-associated malignancies including NPC (32, 33, 34), hypopharyngeal carcinomas (35), Hodgkin lymphomas (36, 37), and non-Hodgkin lymphomas of the gastrointestinal tract (38) for research purposes. However, these studies used different methodical approaches to determine the presence of EBV. Several studies used a single-copy EBV target, for instance, EBNA-1 for the detection of EBV DNA in the tumor sections (32, 34). Because every EBV genome carries up to 11 copies of the BamHI W repeat (39, 40), the use of this fragment as target in the PCR screening definitely increased the sensitivity of the assay. In addition, our PCR was followed by an EIA using alkaline phosphatase (AP) for the detection of the PCR products. AP has a large dynamic range of measurement that enables the distinction between weak and strong positive cases.
This screening strategy is very interesting for testing large groups of tumors of types that have a relatively low association with EBV. Three to 6 times as many samples can be tested by PCR-EIA for the presence of EBV-DNA as compared with EBER1/2-RISH in the same time period. EBV-negative cases with proper DNA quality for PCR amplification do not require further testing by EBER1/2-RISH (68% of the cases). Even when additional DNA purification and EBER1/2-RISH has to be performed on a subset of these cases (total reduction in EBER1/2-RISH to be performed 85%), a larger group can be tested and evaluated in a much shorter time frame. In addition, interpretation of EBER1/2-RISH requires trained personnel, whereas PCR-EIA can be interpreted relatively easily.
In conclusion, we showed that the sensitive BamHI W PCR-EIA is a rapid and reliable screening protocol for the detection of EBV in gastric adenocarcinomas. This approach reduced the amount of samples to be tested by the laborious EBER1/2-RISH with 85%. Only samples with poor DNA quality or with high EBV-PCR hybridization signals require testing for EBV presence in the tumor cells by EBER1/2-RISH. Samples with weak hybridization signals most likely represent cases with EBV-positive infiltrate. Using this protocol we are currently screening a large study group of gastric carcinomas to determine epidemiological and clinicopathological characteristics of EBV-carrying gastric carcinomas in the Netherlands.
References
International Agency for Research on Cancer. Epstein-Barr virus and Kaposi sarcoma herpesvirus/human herpes virus 8.In: IARC monographs on the evaluation of carcinogenic risks to humans. Vol. 70. Lyon, France: International Agency for Research on Cancer; 1997.
Shibata D, Tokunaga M, Uemura Y, Sato E, Takana S, Weiss LM . Association of Epstein-Barr virus with undifferentiated gastric carcinomas with intense lymphoid infiltration. Lymphoepithelioma-like carcinoma. Am J Pathol 1991; 139(3): 469–474.
Oda K, Tamaru J, Takenouchi T, Mikata A, Nunomura M, Saitoh N, et al. Association of Epstein-Barr virus with gastric carcinoma with lymphoid stroma. Am J Pathol 1993; 143(4): 1063–1071.
Nakamura S, Ueki T, Yao T, Ueyama T, Tsuneyoshi M . Epstein-Barr virus in gastric carcinoma with lymphoid stroma. Special reference to its detection by the polymerase chain reaction and in situ hybridization in 99 tumors including a morphologic analysis. Cancer 1994; 73(9): 2239–2249.
Yamamoto N, Tokunaga M, Uemura Y, Tanaka S, Shirahama H, Nakamura T, et al. Epstein-Barr virus and gastric remnant cancer. Cancer 1994; 74(3): 805–809.
Baas IO, van Rees BP, Musler A, Craanen ME, Tytgat GNJ, van den Berg FM, et al. Helicobacter pylori and Epstein-Barr virus infection and the p53 tumour suppressor pathway in gastric stump cancer compared with carcinoma in the non-operated stomach. J Clin Pathol 1998; 51: 662–666.
Shibata D, Weiss LM . Epstein-Barr virus-associated gastric adenocarcinoma. Am J Pathol 1992; 40: 769–774.
Rowlands DC, Ito M, Magham DC, Reynolds G, Herbst H, Hallissey MT, et al. Epstein-Barr virus and carcinomas: rare association of the virus with gastric adenocarcinomas. Br J Cancer 1993; 68: 1014–1019.
Tokunaga M, Uemura Y, Tokudome T, Ishidate T, Masuda H, Okazaki E, et al. Epstein-Barr virus related gastric cancer in Japan: a molecular patho-epidemiological study. Acta Pathol Jpn 1993; 43(10): 574–581.
Fukayama M, Hayashi Y, Iwasaki Y, Chong J, Ooba T, Takizawa T, et al. Epstein-Barr virus-associated gastric carcinoma and Epstein-Barr virus infection of the stomach. Lab Invest 1994; 71(1): 73–81.
Imai S, Koizumi S, Sugiura M, Tokunaga M, Uemura Y, Yamamoto N, et al. Gastric carcinoma: monoclonal epithelial malignant cells expressing Epstein-Barr virus latent infection protein. Proc Natl Acad Sci U S A 1994; 91(19): 9131–9135.
Sugiura M, Imai S, Togunaga M, Koizumi S, Uchizawa M, Okamoto K, et al. Transcriptional analysis of Epstein-Barr virus gene expression in EBV-positive gastric carcinoma: unique viral latency pattern in the tumour cells. Br J Cancer 1996; 74(4): 625–631.
zur Hausen A, Brink AA, Craanen ME, Middeldorp JM, Meijer CJ, van den Brule AJ . Unique transcription pattern of Epstein-Barr virus (EBV) in EBV-carrying gastric adenocarcinomas: expression of the transforming BARF1 gene. Cancer Res 2000; 60(10): 2745–2748.
Ambinder RF, Mann RB . Epstein-Barr encoded RNA. in situ hybridization: diagnostic applications. Hum Pathol 1994; 25: 602–605.
Hamilton Dutoit SJ, Pallesen G . Detection of the Epstein-Barr virus small RNAs in routine paraffin sections using non-isotopic RNA/RNA in situ hybridization. Histopathology 1994; 25 101–111.
Gulley ML . Molecular diagnosis of Epstein-Barr virus-related diseases. J Mol Diagn 2001; 3(1): 1–10.
Komano J, Maruo S, Kurozumi K, Oda T, Takada K . Oncogenic role of Epstein-Barr virus-encoded RNAs in Burkitt's lymphoma cell line Akata. J Virol 1999; 73: 9827–9831.
Clemens MJ . The small RNAs of Epstein-Barr virus. Mol Biol Rep 1993; 17: 81–92.
Gulley ML, Raab-Traub N . Detection of Epstein-Barr virus in human tissues by molecular genetic techniques. Arch Pathol Lab Med 1993; 117: 111–120.
de Bruin PC, Jiwa NM, van der Valk P, van Heerde P, Gordijn R, Ossenkoppele GJ, et al. Detection of Epstein-Barr virus nucleic acid sequences and protein in nodal T cell lymphomas: relation between latent membrane protein-1 and clinical course. Histopathology 1993; 23: 509–518.
de Bruin PC, Jiwa NM, Oudejans JJ, Radaszkiewicz T, Meijer CJ . Epstein-Barr virus in primary gastrointestinal T cell lymphomas. Association with gluten-sensitive enteropathy, pathological features, and immunophenotype. Am J Pathol 1995; 146(4): 861–867.
Bonenkamp JJ, Hermans J, Sasako M, van de Velde CJH, for the Dutch Gastric Cancer Group. Extended lymph-node dissection for gastric cancer. N Engl J Med 1999; 340: 908–914.
Maniatis T, Fritsch EF, Sambrook J . Molecular cloning: a laboratory manual. Cold Spring Harbor, New York: Cold Spring Harbor Laboratory; 1982.
de Roda Husman AM, Walboomers JMM, van den Brule AJC, Meijer CJLM, Snijders PJF . The use of general primer GP5 and GP6 elongated at their 3′ ends with adjacent highly conserved sequences improves human papillomavirus detection by polymerase chain reaction. J Gen Virol 1995; 76 412–417.
Jiwa NM, Kanavaros P, van der Valk P, Walboomers JM, Horstman A, Vos W, et al. Expression of c-myc and bcl-2 oncogene products in Reed-Sternberg cells independent of the presence of Epstein-Barr virus. J Clin Pathol 1993; 46: 211–217.
Jacobs MV, van den Brule AJC, Snijders PJF, Helmerhorst T, Meijer CJLM, Walboomers JMM . A non-radioactive PCR enzyme immunoassay enables a rapid detection of HPV 16 and 18 in cervical smears after GP5+/GP6+ PCR. J Med Virol 1996; 49: 223–229.
Jiwa NM, Kanavaros P, de Bruin PC, van der Valk P, Horstman A, Vos W, et al. Presence of Epstein-Barr virus harbouring small and intermediate-sized cells in Hodgkin's disease. Is there a relationship with Reed-Sternberg cells? J Pathol 1993; 170: 129–136.
Brink AA, ten Berge RL, van den Brule AJ, Willemze R, Chott A, Meijer CJ . Epstein-Barr virus is present in neoplastic cytotoxic T cells in extranodal, and predominantly in B cells in nodal T non-Hodgkin lymphomas. J Pathol 2000; 191(4): 400–406.
Chapel F, Fabiani B, Davi F, Raphael M, Tepper M, Champault G, et al. Epstein-Barr virus and gastric carcinoma in Western patients: comparison of pathological parameters and p53 expression in EBV-positive and negative tumors. Histopathology 2000; 36(3): 252–261.
Galetsky SA, Tvetnov VV, Land CE, Afanasieva TA, Petrovichev NN, Gurtsevitch VE, et al. Epstein-Barr virus associated gastric cancer in Russia. Int J Cancer 1997; 73(6): 786–789.
Satoh Y, Takasaka N, Hoshikawa Y, Osaki M, Ohfuji S, Ito H, et al. Pretreatment with restriction enzyme or bovine serum albumin for effective PCR amplification of Epstein-Barr virus DNA in DNA extracted from paraffin-embedded gastric carcinoma tissue. J Clin Microbiol 1998; 36(11): 3423–3425.
Popat SR, Gunnar Liavaag P, Morton R, McIvor N, Irish JC, Freeman JL . Epstein-Barr virus genome in nasopharyngeal carcinomas from New Zealand. Head Neck 2000; 22(5): 505–508.
Tsai S-T, Jin Y-T, Mann RB, Ambinder RF . Epstein-Barr virus detection in nasopharyngeal tissues of patients with suspect nasopharyngeal carcinoma. Cancer 1998; 82: 1449–1453.
Vera Sempere F, Burgos J, Botella MS, Morera C . Comparative analysis of Epstein-Barr virus (EBV) detection by nested PCR and non-isotopic in situ hybridization in nasopharyngeal carcinoma (NPC). Clin Chim Acta 1998; 271(2): 119–132.
Zhou L, Miyagi Y, Hiroshi E, Tanaka Y, Aoki I, Tsukuda M . Evaluation of Epstein-Barr virus infection in hypopharyngeal carcinomas from 37 Japanese patients. Mod Pathol 1998; 11(6): 509–512.
Uhara H, Sato Y, Mukair K, Akao L, Matsuno Y, Furuya S, et al. Detection of Epstein-Barr virus DNA in Reed-Sternberg cells of Hodgkin's disease using the polymerase chain reaction and in situ hybridization. Jpn J Cancer Res 1990; 81(3): 272–278.
Weiss LM, Chen YY, Liu XF, Shibata D . Epstein-Barr virus and Hodgkin's disease: a correlative in situ hybridization and polymerase chain reaction study. Am J Pathol 1991; 139(6): 1259–1265.
Yang W-I, Cho M-S, Tomita Y, Ohsawa M, Aozasa K . Epstein-Barr virus and gastrointestinal lymphomas in Korea. Yonsei Med J 1998; 39(3): 268–276.
Baer R, Bankier AT, Biggin MD, Deininger PL, Farrell PJ, Gibson TJ, et al. DNA sequence and expression of the B95–8 Epstein-Barr virus genome. Nature 1984; 310: 207–211.
Jones MD, Griffin BE . Clustered repeat sequences in the genome of Epstein-Barr virus. Nucleic Acids Res 1983; 11: 3919–3937.
Acknowledgements
The authors wish to thank Prof. J.H.J.M van Krieken for critically reviewing the manuscript and Mrs. M.J.M. van Zijp and Ms. A. Groothuis for excellent technical assistance.
Author information
Authors and Affiliations
Corresponding author
Additional information
This work was financially supported by Grant VU99-1990 of the Dutch Cancer Society.
Rights and permissions
About this article
Cite this article
van Beek, J., zur Hausen, A., Kranenbarg, E. et al. A Rapid and Reliable Enzyme Immunoassay PCR-Based Screening Method to Identify EBV-Carrying Gastric Carcinomas. Mod Pathol 15, 870–877 (2002). https://doi.org/10.1097/01.MP.0000024147.43288.B1
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1097/01.MP.0000024147.43288.B1
Keywords
This article is cited by
-
Prognostic significance of Epstein-Barr virus infection in gastric cancer: a meta-analysis
BMC Cancer (2015)
-
Association between Epstein-Barr Virus Infection and Chemoresistance to Docetaxel in Gastric Carcinoma
Molecules and Cells (2011)
-
Epstein-Barr virus-specific methylation of human genes in gastric cancer cells
Infectious Agents and Cancer (2010)
-
Meta-analysis of the relationship between Epstein-Barr virus infection and clinicopathological features of patients with gastric carcinoma
Science China Life Sciences (2010)
-
High levels of Epstein–Barr virus DNA in latently infected gastric adenocarcinoma
Laboratory Investigation (2009)
