
Abstract
Pleomorphic rhabdomyosarcoma (PRMS) is a rare and controversial tumor of skeletal muscle phenotype. Diagnostic criteria for PRMS by combined histology and currently available immunohistochemistry have not been clearly defined. We report 38 pleomorphic rhabdomyosarcomas in adults, explore morphologic variants, and discuss our experience with both specific and nonspecific skeletal muscle markers in these tumors. Clinical data, morphology, and immunohistochemistry were reviewed. Electron microscopy was performed. Of 38 cases, there were 28 males and 10 females. Patient ages ranged from 21 to 81 years (median = 54 y; mean = 51 y). Tumors were located in the lower extremity (n = 18), abdomen/retroperitoneum (n = 6), chest/abdominal wall (n = 5), spermatic cord/testes (n = 4), upper extremity (n = 3), and one each in the mouth and orbit. Tumor sizes ranged from 1.5 to 15.0 cm (mean = 7.3 cm; median = 6.8 cm). The cases were divided into three variants, each with large, atypical, pleomorphic polygonal rhabdomyoblasts (PRMB) with abundant eosinophilic cytoplasm in varying numbers and different morphologic backgrounds of round or spindled rhabdomyoblasts (RMB).1. Classic PRMS: Predominantly atypical PRMB in sheets (n = 8).2. Round cell PRMS: Clusters of PRMB throughout the tumor with a background of slightly atypical, medium-sized, round, blue RMB (n = 13).3. Spindle cell PRMS: Scattered PRMB in a predominance of atypical spindled RMB arranged in a storiform growth pattern (n = 17).Immunohistochemistry revealed the following: myoglobin (37/38), MyoD1 (19/36), skeletal muscle myogenin (myf4; 19/34), fast skeletal muscle myosin (4/5), desmin (36/38), muscle-specific actin (MSA; 25/35), smooth muscle actin (SMA; 15/33), and muscle specific myogenin (myf3; 25/35). Immunohistochemistry was supportive of skeletal muscle differentiation with at least one positive skeletal muscle-specific marker (myoglobin, MyoD1, fast skeletal muscle myosin, or myf4). In addition, all cases had some positivity for nonspecific muscle markers (desmin, MSA, SMA, myf3). Electron microscopy (EM), performed on eight selected cases from all three morphologic groups, demonstrated definitive skeletal muscle differentiation in all cases. Follow-up, available on 30 (79%) cases, revealed that 70% of patients died of disease (mean 20 months, range 1 month-108 months), 3% were alive with disease at 12 months (n = 1); and 27% had no evidence of disease (mean, 83 mo; range, 18 to 108 mo). PRMS, a tumor of predominantly middle-aged adult males in the lower extremity, can be diagnosed by the morphologic presence of scattered PRMB with immunohistochemical evidence of at least one skeletal muscle-specific marker. There are three morphologic variants of PRMS. The appropriate diagnosis of PRMS is significant as it is a high-grade sarcoma, with an aggressive clinical course.
Similar content being viewed by others
INTRODUCTION
Rhabdomyosarcoma is rare in adults (1, 2). The most common subtype of rhabdomyosarcoma to occur in adults, however, is the pleomorphic type. Other rhabdomyosarcoma subtypes that occur in adults include the predominantly juvenile alveolar (3, 4) and embryonal (2, 5), the spindle cell subtype of embryonal (6), and the rare adult clear cell and sclerosing pseudovascular subtypes (4). Several studies have addressed PRMS during the past two decades using immunohistochemical techniques, but only on small numbers of tumors. The latest cytoskeletal and noncytoskeletal muscle markers have not yet been fully explored in these tumors. This study defines the clinicopathologic features of a large group of pleomorphic rhabdomyosarcomas, emphasizes morphologic variants, and incorporates the most recent skeletal muscle immunohistochemical markers.
MATERIALS AND METHODS
The records from the Soft Tissue Registry of the Armed Forces Institute of Pathology in Washington, DC from 1970 and from the University of Jena, Germany (see Acknowledgments) were reviewed for cases coded as pleomorphic rhabdomyosarcoma in which the patient was >20 years old. None of the cases had been previously coded as malignant fibrous histiocytoma.At least 123 cases were initially evaluated. Morphologically, only cases showing at least scattered, large, atypical pleomorphic tumor cells with abundant eosinophilic cytoplasm were accepted for further diagnostic studies. Immunohistochemical stains were performed on 81 cases with available blocks or unstained slides, including antibodies to desmin, myoglobin, myf4, myf3, fast myosin, MyoD1, vimentin, keratin, S-100 protein, CD 31, CD 34, EMA, SMA, and MSA. MyoD1, myf3, myf4, and S100 protein are nuclear stains; S100 may also be cytoplasmic diagnostically. Myoglobin was required to show central tumor cell positivity and not just edge effect or nonspecific, tea-colored–like staining. Thirty-eight cases with immunohistochemistry were eventually included in this study. Thirty-two of these cases were studied by the avidin biotin complex system with a diaminobenzoic acid chromogen; six cases were studied by the alkaline-phosphatase-antialkaline phosphatase method (APAAP). Appropriate positive and negative controls were used throughout. The primary antibodies used and the dilutions and sources are listed in Table 1.
Thirty-eight cases fulfilled our PRMS criteria. All cases better diagnosed as other tumors, including other pleomorphic sarcomas such as malignant fibrous histiocytoma (otherwise considered pleomorphic sarcoma, not otherwise specified), leiomyosarcoma, and malignant Triton tumor, were excluded from this study.
Electron microscopy (see Acknowledgments) was performed on eight selected paraffin blocks representing all three PRMS subtypes, including Cases 2, 4, 11, 12, 17, 28, 30, and 38. Specimens were reclaimed from paraffin, rehydrated, and fixed in glutaraldehyde, followed by a secondary osmium tetroxide fixation and embedment in epoxy. Thin sections were double stained with uranyl acetate and lead citrate and viewed on a JEOL 100S electron microscope.
Follow-up information was obtained on 30 (79%) cases from the patient’s medical records, the patient’s physicians, and occasionally, the patients themselves.
RESULTS
Clinical information and follow-up are presented in Table 2. The 38 tumors occurred in 28 men and 10 women. Patient ages ranged from 21 to 81 years (median = 54 years; mean = 51 years). Tumors were located in the lower extremity (n = 18), abdomen/retroperitoneum (n = 6), chest/abdominal wall (n = 5), spermatic cord/testes (n = 4), upper extremity (n = 3), and one each in the mouth and orbit. Most patients presented with a rapidly growing, painful swelling. Two cases were described as trauma related.
Grossly, the tumors were well circumscribed, lobulated, and often surrounded by a fibrous pseudocapsule. The cut surface was gray, white, and firm with variable amounts of hemorrhage. Necrosis was identified grossly in several cases. One case, arising in the scrotum and testes, had two separate tumor nodules. Tumor sizes ranged from 1.5 to 15.0 cm, with a mean of 7.3 cm and a median of 6.8 cm.
Morphology revealed large atypical, polygonal, pleomorphic rhabdomyoblasts in all cases. These cells varied from strap-like and spindled to racquet-shaped or multinucleated with vesicular nuclei, prominent nucleoli, and abundant eosinophilic cytoplasm. Cross striations were identified in only one case. The background rhabdomyoblasts varied from sheets of purely polygonal large- to medium-sized cells to round or epithelioid cells, to sheets of atypical, pleomorphic spindled cells in a storiform growth pattern. The average mitotic count was 16/10HPF. Geographic necrosis was present in 33/38 cases. Five cases exhibited perivascular tumor sparing in areas of abundant necrosis.
The cases were divided into three morphologic variants presented in Table 3. The large atypical polygonal pleomorphic cells (PRMS) and background cells (RMS) were determined to be rhabdomyoblasts by skeletal muscle-specific markers, as outlined in the immunohistochemical results below (Fig. 1):
-
1
1. Classic PRMS: sheets of large, atypical and often multinucleated polygonal pleomorphic rhabdomyoblasts (PRMB; n = 8; Fig. 2).
FIGURE 2 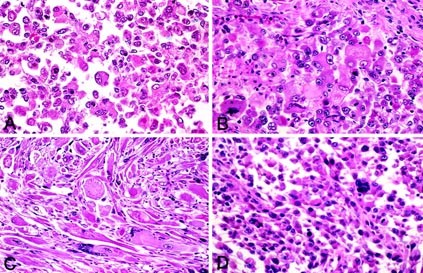
Representative examples of Type 1 (classic variant) pleomorphic rhabdomyosarcoma, with sheets of atypical, bizarre, large, polygonal pleomorphic rhabdomyoblasts with abundant eosinophilic cytoplasm (A–D).
-
2
2. Round cell PRMS: clusters of large, atypical PRMB throughout the lesion with a background of slightly pleomorphic medium-sized, round, blue rhabdomyoblasts (RMB; n = 13; Fig. 3).
FIGURE 3 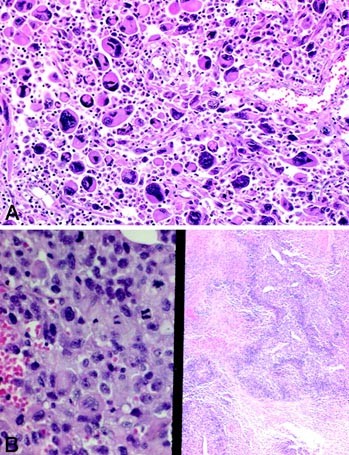
Representative examples of Type 2 (round-cell variant) pleomorphic rhabdomyosarcoma, with scattered pleomorphic rhabdomyoblasts among a background of medium sized, slightly angulated round-to-epithelioid rhabdomyoblasts (A). Higher magnification of this variant (B, left); note the geographic necrosis (common to all morphologic variants; B, right).
-
3
3. Spindle cell PRMS: a predominance of pleomorphic spindled RMB arranged in a storiform growth pattern with scattered large PRMB (n = 17; Fig. 4).
FIGURE 4 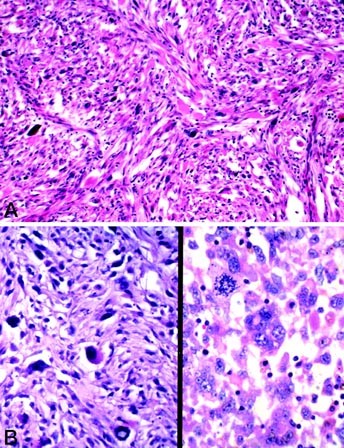
Representative examples of Type 3 (spindle cell variant) pleomorphic rhabdomyosarcoma, with scattered large polygonal pleomorphic rhabdomyoblasts and a spindled, storiform background of rhabdomyoblasts (A–B, left); the atypia, atypical mitoses, and bizarre giant cells are common to all variants (B, right).




An example of a pleomorphic rhabdomyosarcoma (classic variant; left) and diffusely positive desmin reactivity (right; A); myoglobin positivity (B); MyoD1 (nuclear, left) and fast myosin (cytoplasmic, BI) positivity (C); and myogenins myf 3 (nuclear, left) and myf4 (nuclear, BI) positivity (D).
Immunohistochemical results are presented in Table 4. The immunoprofiles established all PRMS cases as having skeletal muscle phenotype, since the large polygonal PRMB and the background RMB were positive for at least one skeletal muscle-specific marker. The immunohistochemical results include the following: myoglobin (37/38; 97%), myoD1 (19/36; 53%), skeletal muscle myogenin (myf4; 19/34; 56%), fast skeletal muscle myosin (4/5; 80%), desmin (36/38; 95%), MSA (25/35; 71%), SMA (15/33; 45%), and myf3 (25/35; 71%).
The two desmin-negative cases were positive for myoglobin and nuclear MyoD1 and one additionally for myf4. The cases with purely cytoplasmic positivity for MyoD1 (n = 14) were considered negative for this marker, although they were included in the group of PRMS by other skeletal muscle-specific markers and electron microscopy. The one case that was negative for myoglobin was positive for myogenin (myf4) and the actins (SMA and MSA), myf3, and desmin. Skeletal muscle-specific markers included myoglobin (if interpreted carefully, see Materials and Methods), MyoD1 (nuclear only), myf4, and fast myosin. Of the skeletal muscle-specific markers, 11 cases had myoglobin-only positive, 1 had myf4-only positive, 13 cases had two skeletal muscle-specific markers positive, 11 cases had three skeletal muscle-specific markers positive, and 2 cases had all four skeletal muscle-specific markers positive. Antibody to fast skeletal muscle myosin was available on only 5 cases.
Electron microscopy was performed from paraffin-embedded tissue on eight cases from this study, including at least one case from each morphologic subtype. The six electron microscopy cases from Group three (spindle cell PRMS) revealed findings as follows: large aggregates of Z-band material and thick myosin and thin actin filaments (four cases), haphazardly arranged sarcomeres with M bands (one case), and ribosome-myosin complexes (one case). Of the remaining three cases from Categories 1 and 2, all showed evidence of skeletal muscle differentiation as thick myosin filaments, myosin-ribosome complexes, or rudimentary sarcomere formation (Fig. 5).

Representative electron microscopy of a pleomorphic rhabdomyosarcoma that confirms skeletal muscle phenotype by thick and thin filaments forming rudimentary Z bands.
Follow-up, available on 30 (79%) cases, revealed that 70% patients died of disease (mean, 20 mo; range, 1 to 108 mo), 3% were alive with disease at 12 months (n = 1); and 27% had no evidence of disease (mean, 83 months; range, 18 to 108 mo). Forty-five percent of patients had their first known local recurrence within a mean of 9 months (range, 2 to 24 mo), with up to two recurrences reported. Fifty-five percent of patients had known metastases within a mean follow-up of 15 months (range, 36 mo). Follow-up for the three morphologic variants was as follows: Type I (Classic PRMS): 5/8 died of disease (DOD; 63%; average time 29 mo), 2/8 had no evidence of disease (NED; 25%). Type II (round cell PRMS): 10/13 DOD (76%; average time, 21 mo), 3/13 NED (23%). And Type III (spindle cell PRMS): 9/17 DOD (53%; average time, 33 mo), 4/17 NED (24%).
DISCUSSION
Historically, Stout first introduced pleomorphic rhabdomyosarcoma (PRMS) into the literature in 1946 (7) as “classical” rhabdomyosarcoma. For the next three decades, the diagnosis was based on morphology alone and probably included other sarcomas (including malignant fibrous histiocytoma and pleomorphic leiomyosarcoma), with cross and longitudinal striations (3, 5, 7, 8, 9, 10, 11, 12, 13). In 1958, Horn and Enterline outlined four subtypes of rhabdomyosarcoma and called the classical ones “pleomorphic rhabdomyosarcoma.” The popularity of PRMS waxed and waned with the introduction of electron microscopy (14, 15, 16, 18, 19) and the entity of malignant fibrous histiocytoma (20), respectively. Immunohistochemical antibodies were applied to these tumors in the early 1980s, predominantly using myoglobin, desmin, creatinine kinase subunit M, and various actins to detect skeletal muscle differentiation (18, 19, 21, 22, 23, 24, 25, 26). With the exception of myoglobin, a protein found late in embryonic muscle development and requiring experienced interpretation, all other antibodies were found to be nonspecific for skeletal muscle phenotype. In the most recent decade, several series have used an immunohistochemical approach to identify PRMS (15, 17, 27, 28, 29, 30, 31). In 1993, fast myosin, a skeletal muscle-specific marker, was added to the repertoire for PRMS (15). MyoD1, the product of a gene activated early in myogenesis, was discussed as a skeletal muscle-specific marker but was not applied to pleomorphic rhabdomyosarcoma until 1995 (31, 32, 33). Myf4, a skeletal muscle-specific myogenin, has only been studied on four cases of PRMS in the literature (27, 34). The current study reports 38 adult cases of PRMS, from the 1970s to the present, diagnosed by using a current morphologic and immunohistochemical approach, the latter aforementioned specific and nonspecific striated muscle markers.
Clinically, our study reveals that pleomorphic rhabdomyosarcoma is an aggressive sarcoma, arising predominantly in the extremities of adult males with a mean age of 49 years. Our clinicopathologic findings are similar to those cases reported in the literature (3, 5, 7, 8, 9, 10, 11, 12, 13, 15, 21, 23, 25, 27, 29, 31. The overall prognosis in the literature for PRMS is poor, with survival rates of 12.5 to 50% of 1-year to 20-month disease-free survival, (3, 5, 7, 8, 9, 10, 11, 13, 15, 17, 18, 29) similar to our 27% rate of 5-year disease free survival. On the basis of our follow-up, 72% of patients died within 2.0 years of diagnosis. Comparatively, storiform pleomorphic type of malignant fibrous histiocytoma, another pleomorphic sarcoma, has overall 5-year survival rates varying from 36 to 50% (35, 36). Pleomorphic liposarcoma has a 5-year survival of 56% (37), and deep extremity leiomyosarcomas a 64% 5-year survival rate (38). However, these latter tumors were not all pleomorphic subtype. Therefore, pleomorphic rhabdomyosarcoma in adults has a significantly worse prognosis than that for other pleomorphic sarcomas. Thus, the correct diagnosis of pleomorphic rhabdomyosarcoma is important.
Morphologically, all of our pleomorphic rhabdomyosarcomas were composed of large, atypical, polygonal pleomorphic rhabdomyoblasts with abundant eosinophilic cytoplasm. These large rhabdomyoblasts are often arranged in clusters, sheets, or scattered individual cells. Atypical, vesicular nuclei with prominent nucleoli predominate. The rhabdomyoblasts in the background that surround the large, pleomorphic rhabdomyoblasts vary from round to spindled. The morphologic features of the large atypical rhabdomyoblasts in pleomorphic rhabdomyosarcoma have been previously described by Stout (7) and others (3, 5, 8, 9, 10, 11, 12, 13, 15, 21, 29, 31). However, with the exception of one group (29), who described a background of larger round cells, presumably rhabdomyoblasts, mainly only spindled and polygonal background cells have been previously recognized in the literature (15, 16, 17, 18, 29). The above morphologic characteristics of the cases in the current study, with atypical large polygonal pleomorphic rhabdomyoblasts and two additional types of background rhabdomyoblasts, distinguish our proposed recognized morphologic variants of pleomorphic rhabdomyosarcoma as classic, round cell, and spindle cell variants, respectively.
The immunoprofiles of our PRMS reveal that all cases have at least one skeletal muscle-specific marker positive (myoglobin, nuclear MyoD1, nuclear myf4, and fast myosin) in addition to positivity for nonspecific myoid markers, including desmin, myogenin (myf3) or MSA. Although not enough material was available to test all of the antibodies on every case, myoglobin (95%) and fast skeletal muscle myosin (80%) appeared to be the most sensitive markers for PRMS in our series. These findings compare similarly with those of myoglobin and fast skeletal muscle myosin on PRMS in the literature (15, 23, 28, 29). Although myoglobin is often a difficult stain to interpret due to its high background, in this study, we found myoglobin to be highly sensitive for these poorly differentiated rhabdomyosarcomas, if interpreted carefully. We required tumor cell cytoplasm within the center of the tumor (not just edge effect) and stronger than just background “tea-color” to call a tumor positive. Fast skeletal muscle myosin was only studied on five of our cases; additional cases would need to be examined with this marker for further data.
MyoD1 and the myogenins (including myf4 and myf3) belong to a family of closely related genes encoding a series of DNA binding proteins that are thought to control myogenesis (27, 32, 34). MyoD1 and myf4 were less sensitive for PRMS, in our experience. However, we also used strict criteria for MyoD1, i.e., requiring nuclear reactivity. Interestingly, fourteen additional PRMS cases, included by other skeletal muscle specific markers and electron microscopy, showed only cytoplasmic positivity for MyoD1. If these fourteen were counted as true positives, it would increase the sensitivity of MyoD1 from 51 to 89%. Cytoplasmic positivity therefore should be used as a supportive but nonspecific finding for PRMS. The sensitivity of MyoD1 is as high as 90% in the literature, yet only ten cases have been tested (27, 31). Myf4, myogenin, a supportive skeletal muscle marker, was only 53% sensitive for PRMS, in our experience. The experience in the literature, on only four cases of pleomorphic rhabdomyosarcoma, is 75% specificity (27). Myf4, however, has been previously identified as being more specific for skeletal muscle differentiation than myf3, in rhabdomyosarcomas in general. In one abstract (39), myf3 also stained 70% of leiomyosarcomas, 67% of malignant fibrous histiocytomas, 100% of Wilm’s tumors, 55% of other small round cell tumors, and 10% of other tumors. Whereas, myf4 was specific for rhabdomyosarcomas with no other staining, except for one case of Wilm’s tumor, of the above mentioned tumors. Further information on myf4, from a recent publication, reveals that it is highly sensitive and specific for rhabdomyosarcoma compared with other pediatric tumors (40).
Electron microscopy, with a minimal criterium of ribosome-myosin complexes, was supportive of skeletal muscle phenotype in all eight cases studied, representing all three of our subtypes of PRMS.
We propose three morphologic variants. All three of these variants of PRMS are required to have at least one skeletal muscle specific marker positive in addition to other muscle markers. Type I or “classic PRMS” is defined morphologically by sheets of large, atypical polygonal, pleomorphic rhabdomyoblasts (PRMB). Our series included 8 such cases. Type II, also termed “round cell PRMS,” was composed morphologically of these large PRMB among medium sized slightly pleomorphic round rhabdomyoblasts. These comprised 13 of our cases. Although one may consider this morphologic variant to have similarities to embryonal rhabdomyosarcoma, there are several reasons why these cases are better classified as the round cell variant of pleomorphic rhabdomyosarcoma. These tumors are all in adults. The round cells are larger than the round cells of embryonal rhabdomyosarcoma, and there are more numerous and more atypical pleomorphic rhabdomyoblasts within these tumors. Solid alveolar rhabdomyosarcoma subtype can be distinguished from this round cell variant of PRMS by its t(2;13) or variant t(1;13) chromosomal translocations. The same PAX7 site on chromosome 1 appears to be involved in the genetic changes of embryonal rhabdomyosarcoma (41). Comparatively, not much is known about the genetics of PRMS. Further genetic studies would aid in our understanding of the relationship of PRMS to other RMS subtypes. Type III, or spindle cell PRMS, accounted for the majority of our cases (n = 17). It was composed of large, atypical pleomorphic rhabdomyoblasts with highly spindled and often storiform backgrounds. These tumors mimic storiform pleomorphic malignant fibrous histiocytoma, yet have large, atypical rhabdomyoblasts and immunophenotypic evidence for skeletal muscle differentiation in polygonal and spindled cells. These cases were highly pleomorphic and could easily be separated from the spindle cell variant of embryonal rhabdomyosarcoma, also described in adults (6).
Our morphologic variants of pleomorphic rhabdomyosarcoma are based on the predominant morphologic pattern and do not differ substantially with respect to prognosis, although there is a slightly worse prognosis for the round cell variant. On the other hand, our numbers are small, and therefore statistically significance cannot be determined. All three groups have relatively poor prognosis. We believe that it is important to recognize this morphologic spectrum of pleomorphic rhabdomyosarcoma for diagnostic purposes and in order to help separate pleomorphic rhabdomyosarcoma from other tumors.
Along with interpretation of myogenic immunohistochemical stains, a broad panel of nonmyoid markers should be evaluated to rule out poorly differentiated carcinomas, rhabdoid tumors, Triton tumors, and malignant melanoma. Although rhabdomyosarcomas, like other sarcomas, may occasionally express keratin, lack of epithelial markers may help to separate PRMS from carcinomas and extrarenal rhabdoid tumors. Lack of focal S100 protein and GFAP and presence of myoid markers in the background spindled rhabdomyoblasts help to identify the spindled component of spindle cell PRMS as separate from the malignant peripheral nerve sheath tumor component of malignant Triton tumor. Malignant melanoma often has diffuse positivity for S100 protein as well as possible positivity for melanocytic markers, including HMB-45, melan-A, and tyrosinase.
Malignant fibrous histiocytoma (MFH) (sometimes considered “pleomorphic sarcoma, not otherwise specified”) and pleomorphic leiomyosarcoma (LMS) may also considered in the differential diagnosis. MFH, although known to occasionally express both desmin and MSA (42) should not express other specific skeletal muscle markers, such as MyoD1, fast skeletal muscle myosin, myf4, or myoglobin. LMS, a myoid tumor with desmin expression, morphologically has intersecting fascicles, lacks the presence of large polygonal rhabdomyoblasts, and also does not express skeletal muscle specific markers.
In conclusion, this study reviews 38 cases of PRMS. PRMS occurs most often in the extremities of adult males and typically follows an aggressive clinical course with an overall poor prognosis. Three morphologic variants have been identified based on the presence of large, polygonal, atypical pleomorphic rhabdomyoblasts with two additional shapes of background rhabdomyoblasts: classic, round cell, and spindle cell pleomorphic rhabdomyosarcoma, respectively. Immunohistochemical analysis should include both specific skeletal muscle markers and nonspecific myoid markers. There may be a spectrum of differentiation in PRMS, as evidenced by variable myoregulatory protein expression, which does not appear to be detected on a purely morphologic basis.
References
Hollowood K, Fletcher CDM . Rhabdomyosarcoma in adults. Semin Diag Pathol 1994; 11: 47–57.
Miettinen M . Rhabdomyosarcoma in patients older than 40 years of age. Cancer 1988; 62: 2060–2065.
Horn RC, Enterline HT . Rhabdomyosarcoma: a clinicopathological study and classification of 39 cases. Cancer 1958; 11: 181–197.
Mentzel T, Katenkamp D . Sclerosing, pseudovascular rhabdomyosarcoma in adults. Clinicopathological and immunohistochemical analysis of three cases. Virchows Arch 2000; 436: 305–311.
Albores-Saavedra J, Martin RG, Smith JL . Rhabdomyosarcoma. A study of 35 cases. Ann Surg 1963; 157: 186–197.
Rubin BP, Hasserjian RP, Singer S, Janecka I, Fletcher JA, Fletcher CDM . Spindle cell rhabdomyosarcoma (so-called) in adults: report of two cases with emphasis on differential diagnosis. Am J Surg Pathol 1998; 22: 459–464.
Stout AP . Rhabdomyosarcoma of skeletal muscles. Ann Surg 1946; 123: 447–472.
Ariel IM, Briceno M . Rhabdomyosarcoma of the extremities and trunk: analysis of 150 patients treated by surgical resection. J Surg Oncol 1975; 7: 269–287.
Keyhani A, Booher RJ . Pleomorphic rhabdomyosarcoma. Cancer 1968; 22: 956–967.
Linsheid RL, Soule EH, Henderson ED . Pleomorphic rhabdomyosarcoma of the extremities and limb girdles. A clinicopathologic study. J Bone Joint Surg 1965; 47A: 715–726.
Pack GT, Eberhart WF . Rhabdomyosarcoma of skeletal muscle. Report of 100 cases. Surgery 1952; 32: 1023–1064.
Patton RB, Horn RC . Rhabdomyosarcoma. Clinical and pathological features and comparison with human fetal and embryonal skeletal muscle. Surgery 1962; 52: 572–584.
Phelan JT, Juardo J . Rhabdomyosarcomas. Surgery 1962; 52: 585–591.
Erlandson RA . The ultrastructural distinction between rhabdomyosarcoma and other undifferentiated “sarcomas.” Ultrastruct Pathol 1987; 11: 83–101.
Gaffney EF, Dervan PA, Fletcher CDM . Pleomorphic rhabdomyosarcoma in adulthood. Analysis of 11 cases with definition of diagnostic criteria. Am J Surg Pathol 1993; 17: 601–609.
Reddick RL, Michelitch H, Triche TJ . Malignant soft tissue tumors (malignant fibrous histiocytoma, pleomorphic liposarcoma, and pleomorphic rhabdomyosarcoma): an electron microscopic study. Hum Pathol 1979; 10: 327–343.
Schurch W, Begin LR, Seemayer TA, Legace R, Boivin JC, Lamoureux C, et al. Pleomorphic soft tissue myogenic sarcomas of adulthood. A reappraisal in the mid-1990’s. Am J Surg Pathol 1996; 20 (2): 131–147.
Skalli O, Gabbiani G, Babai F, Seemayer TA, Pizzolato G, Schurch W . Intermediate filament proteins and actin isoforms as markers for soft tissue tumor differentiation and origin. Am J Pathol 1988; 130: 515–531.
Weiss LM, Warhol MJ . Ultrastructural distinction between adult pleomorphic fibrous histiocytomas. Hum Pathol 1982; 15: 1025–1033.
Enzinger FM . Malignant fibrous histiocytoma 20 years after Stout. Am J Surg Pathol 1986; 10 (1 Suppl): 43–53.
de Jong ASH, van Kessel-van Vark M, Albus-Lutter CHE . Pleomorphic rhabdomyosarcoma in adults: immunohistochemistry as a tool for its diagnosis. Hum Pathol 1987; 18: 298–303.
de Jong ASH, van Kessel-van Vark M, Albus-Lutter CE, van Raamsdonk W, Voute PA . Myosin and myoglobin as tumor markers in the diagnosis of rhabdomyosarcoma. A comparative study. Am J Surg Pathol 1984; 8: 521–528.
Leader M, Patel J, Collins M, Henry K . Myoglobin: an evaluation of its role as a marker of rhabdomyosarcomas. Br J Cancer 1989; 59: 106–109.
Rangdaeng S, Truong LD . Comparative immunohistochemical staining for desmin and MSA. A study of 576 cases. Am J Clin Pathol 1991; 96: 32–45.
Schmidt RA, Cone R, Haas JE, Gown AM . Diagnosis of rhabdomyosarcoma with HHF35, a monoclonal antibody directed against muscle actins. Am J Pathol 1998; 131: 19–28.
Truong LD, Rangdaeng S, Cagle P, Ro JY, Hawkins H, Font RL . The diagnostic utility of desmin. Am J Clin Pathol 1990; 93: 305–314.
Cui S, Hano H, Harada T, Takai S, Masui F, Ushigome S . Evaluation of new monoclonal anti-myoD1 and anti-myogenin antibodies for the diagnosis of rhabdomyosarcoma. Pathol Int 1999; 49: 62–68.
Gruchala A, Niezabitowski A, Wasilewska A, Sikora K, Rys J, Szklarski W, et al. Rhabdomyosarcoma. Morphologic, immunohistochemical and DNA study. Gen Diagn Pathol 1997; 142 (3–4): 175–184.
Ordi J, Stamatakos MD, Tavassoli FA . Pure pleomorphic rhabdomyosarcoma of the uterus. Int J Gynecol Pathol 1997; 16 (4): 369–377.
Suster S, Moran CA, Koss MN . Rhabdomyosarcomas of the anterior mediastinum: report of four cases unassociated with germ cell, teratomatous or thymic carcinomatous components. Hum Pathol 1994; 25: 349–356.
Wesche WA, Fletcher CDM, Dias P, Houghton PJ, Parham DM . Immunohistochemistry of myoD1 in adult pleomorphic soft tissue sarcomas. Am J Surg Pathol 1995; 19 (3): 261–269.
Dias P, Parham DM, Shapiro DN, Tapscott ST, Houghton PJ . Monoclonal antibodies to the myogenic regulatory protein myoD1: epitope mapping a diagnostic utility. Cancer Res 1992; 52: 6431–6439.
Hosoi H, Sugimoto T, Hayashi Y, Inaba T, Honii Y, Monoka H, et al. Differential expression of myogenic regulatory genes myoD1 and myogenin, in human rhabdomyosarcoma sublines. Int J Cancer 1992; 50: 977–983.
Dias P, Chen B, Dilday B, Palmer H, Hosoi H, Singh S, et al. Strong immunostaining for myogenin in rhabdomyosarcoma is significantly associated with tumors of the alveolar subclass. Am J Pathol 2000; 156 (2): 399–408.
Bertoni F, Capanna R, Biagini R, Bacchini P, Guerra A, Ruggieri P, et al. Malignant fibrous histiocytoma of soft tissue. An analysis of 78 cases located and deeply seated in the extremities. Cancer 1985; 56: 356–367.
Pezzi CM, Rawlings MS Jr, Esgro JJ, Pollock RE, Romsdahl MM . Prognostic factors in 227 patients with malignant fibrous histiocytomas. Cancer 1992; 69: 2098–2103.
Chang HR, Hajdu SI, Collin C, Brennan MF . The prognostic value of histologic subtypes in primary extremity liposarcoma. Cancer 1989; 64: 1514–1520.
Gustafson P, Willen H, Baldetorp B, Ferno M, Akerman M, Rydholm A . Soft tissue leiomyosarcoma: a population based epidemiologic and prognostic study of 48 patients, including cellular DNA content. Cancer 1992; 70: 114–119.
Berezowski K, Workman R, Kornstein MJ . Antibodies to myogenic regulatory proteins in the diagnosis of rhabdomyosarcoma [abstract]. Mod Pathol 1998; 11: 7A.
Kumar S, Perlman E, Harris CA, Raffeld M, Tsokos M . Myogenin is a specific marker for rhabdomyosarcoma: an immunohistochemical study in paraffin-embedded tissues. Mod Pathol 2000; 13: 988–993.
Bridge JA, Liu J, Weibolt V, Baker KS, Perry D, Druger R, et al. Novel genomic imbalances in embryonal rhabdomyosarcoma revealed by comparative genomic hybridization and fluorescence in situ hybridization: an intergroup rhabdomyosarcoma study. Genes Chromosomes Cancer 2000; 27: 337–344.
Miettinen M . Immunohistochemistry of soft tissue tumors. Possibilities and limitations in surgical pathology. Pathol Annu 1990; 25 (Pt 1): 1–36.
Acknowledgements
The authors are grateful to Professor Dr. D. Katenkamp, University of Jena, Germany, who kindly gave permission to include cases of his personal consultation files in this study. We also greatly appreciate the excellent technical assistance of Eileen Rusnock, B.A., who performed the electron microscopy for this paper.
Author information
Authors and Affiliations
Corresponding author
Additional information
The opinions and assertions contained herein are the expressed views of the authors and are not to be construed as official or as a reflection of the views of the Departments of the Army or Defense.
Rights and permissions
About this article
Cite this article
Furlong, M., Mentzel, T. & Fanburg-Smith, J. Pleomorphic Rhabdomyosarcoma in Adults: A Clinicopathologic Study of 38 Cases with Emphasis on Morphologic Variants and Recent Skeletal Muscle-Specific Markers. Mod Pathol 14, 595–603 (2001). https://doi.org/10.1038/modpathol.3880357
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.3880357
Keywords
This article is cited by
-
Characterization of a rhabdomyosarcoma reveals a critical role for SMG7 in cancer cell viability and tumor growth
Scientific Reports (2023)
-
Haploinsufficiency of the lysosomal sialidase NEU1 results in a model of pleomorphic rhabdomyosarcoma in mice
Communications Biology (2022)
-
Pleomorphic rhabdomyosarcoma in a young adult harboring a novel germline MSH2 variant
Human Genome Variation (2022)
-
Upfront surgical resection for primary bone tumors: rationale and potential benefits
Surgical and Experimental Pathology (2020)
-
The current landscape of rhabdomyosarcomas: an update
Virchows Archiv (2020)
