
Abstract
Background: Inflammatory myofibroblastic tumor (IMT) is an uncommon tumor of extrapulmonary and pulmonary tissues with an unpredictable clinical course, occasional recurrences, and rare malignant transformation. Clonal abnormalities with rearrangements of chromosome of 2p23 and the ALK gene have been reported in a few cases. The purpose of this study is to investigate whether these are consistent abnormalities among IMTs or represent a distinct subset. Design: Formalin-fixed, paraffin-embedded archival tissue sections from 47 IMTs in 40 patients were immunostained with monoclonal antibodies against ALK and p80. Fluorescence in situ hybridization for ALK rearrangements was done on 22 IMTs from 19 patients. Findings were correlated with clinical features and outcome. Results: ALK positivity was observed in 17 of 47 IMTs (36%) and p80 positivity in 16 of 47 IMTs (34%). Fluorescence in situ hybridization showed ALK rearrangements in nine cases (47%), aneuploidy in three cases (16%), and no rearrangement in seven cases (37%). IMTs with ALK abnormalities by immunohistochemistry and/or fluorescence in situ hybridization originated in the abdomen/pelvis/retroperitoneum, chest, and extremities. The mean age was 6.6 years, with a male/female ratio of 1.3. 64% of patients had no evidence of disease at last follow-up, 45% had one or more recurrences, and 18% displayed histologic evidence of malignant transformation. The IMTs without ALK abnormalities occurred in older children, were more frequent in females, and had fewer recurrences. However, in this group of 40 patients, the differences between the groups with and without ALK abnormalities did not have statistical significance. Aneuploidy without ALK abnormalities was associated with malignant transformation in three of five cases. Conclusions: Abnormalities of ALK and p80 and evidence of chromosomal rearrangements of 2p23 occur in a significant proportion of IMTs. These changes are most frequent in abdominal and pulmonary IMTs in the first decade of life and are associated with a higher frequency of recurrence. These findings confirm the neoplastic nature of a subset IMT with ALK abnormalities and suggest that aneuploid IMT is a subset with more aggressive clinical behavior.
Similar content being viewed by others


INTRODUCTION
Inflammatory myofibroblastic tumor (IMT), a tumor of myofibroblastic spindle cells accompanied by a variable inflammatory infiltrate of plasma cells, eosinophils, and lymphocytes, occurs in pulmonary and extrapulmonary sites, recurs locally, manifests systemic symptoms, and rarely undergoes malignant transformation (1, 2, 3, 4). The search for histologic prognostic features has proven elusive (5, 6). One of the fundamental controversies surrounding IMT is whether it is a neoplasm or a reactive pseudotumorous proliferation (2, 7, 8).
Limited information is available about the cytogenetic findings in IMT. Several case reports and small series have reported karyotypic abnormalities, which are summarized in Table 1 (4, 9, 10, 11, 12, 13). These reports indicate that at least some IMTs have clonal aberrations involving the short arm of chromosome 2 in region p21-p23 and support the contention that IMT is a neoplasm.
Chromosome 2p23 is the site of the human ALK gene, which codes for anaplastic lymphoma kinase (14). ALK is a tyrosine kinase receptor and a member of the insulin growth factor receptor superfamily. Clonal abnormalities of ALK are typically associated with anaplastic large cell lymphoma (ALCL), which has a characteristic translocation t (2;5)(p23;q35) in about 40% of cases (15, 16, 17, 18, 19, 20). Recently variant translocations have been cloned in ALCL (21, 22, 23, 24, 25, 26). The t(2;5) in ALCL results in novel fusion proteins, with constitutive active ALK tyrosine kinase domain. One hypothesis for the pathogenesis of ALCL is that the fusion proteins from the translocations lead to abnormal intracellular phosphorylation and neoplastic transformation.
Recently antibodies to the protein product of the ALK gene have been developed that detect both ALK expression associated with the t(2;5) and other abnormalities that may result in deregulation of the ALK gene (19, 20, 27, 28, 29, 30). The t(2;5) is associated with nuclear and cytoplasmic immunoreactivity for two antibodies, ALK-1, a monoclonal antibody, and p80NPM/ALK, a rabbit polyclonal antibody. Variant translocations involving chromosome 2 and other chromosomes in addition to chromosome 5 are associated with cytoplasmic ALK-1 reactivity in ALCL (19, 25, 28).
The purposes of this study are to further investigate the neoplastic versus pseudotumorous nature of IMT by looking for evidence of chromosome rearrangements of 2p23 with involvement of the ALK gene and to assess whether IMTs with ALK rearrangements or deregulation constitute a distinct clinical and pathological subset.
MATERIALS AND METHODS
Case Selection
Formalin-fixed, paraffin-embedded archival tissue from 47 IMTs in 40 patients was obtained from the consultation and institutional files of the authors. Twenty-six of the cases were previously published (1). Flow cytometric ploidy analysis was previously published for 24 cases (6). Clinical data were obtained by review of medical records or through communication with primary care physicians, surgeons, or referring pathologists. All pathology reports and representative microscopic slides were reviewed. Histologic features of IMT have been previously published (1). Metaphase cytogenetics was performed using standard methods at the time of initial evaluation in two cases, one of which was previously published (9).
Immunophenotypic Studies
Immunohistochemical studies were performed on 47 samples from 40 patients using formalin-fixed, paraffin-embedded tissue sections, a standard avidin-biotin peroxidase complex technique, and an automated immunostainer (Ventana Medical Systems, Tucson, AZ). The polyclonal antibody p80NPM/ALK was obtained from Monosan, Uden, The Netherlands, and the working dilution was 1:60. The monoclonal ALK-1 antibody was provided by David Mason, M.D., Oxford, England, and the working dilution was 1:40 (29, 30, 31). Appropriate positive and negative controls were used. Reactivity was interpreted as positive or negative; positive cases were further scored with 1+ representing less than 25% positivity, 2+ representing 25 to 75% positivity, and 3+ representing more than 75% positivity. Cases with positive staining were assessed as to the localization of staining to cytoplasm, nucleus, or both.
Fluorescence In Situ Hybridization (FISH) for ALK
Nuclei were prepared from paraffin-embedded tissue of 22 samples from 19 patients as described previously (32). Paraffin embedded tissue blocks were sectioned at 50 millimicrons, and 3 to 4 sections were used to prepare nuclei. The sections were deparaffinized twice in xylene for 30 minutes and 10 minutes, followed by rehydration in 100%, 95%, 75%, and 50% ethanol in water for 5 minutes each. Tissue sections were digested with Carlsberg solution (0.1% Sigma protease XXIV, 0.1 m Tris, 0.07 m NaCl, pH 7.2) for 1 to 2 hours at 37°C. The resulting nuclear suspensions were vortexed for 20 minutes and filtered through a 50 micron nylon mesh. The nuclei were washed once in phosphate-buffered saline and fixed in methanol and acetic acid (3:1).
For FISH, the nuclear suspensions were pipetted on Vectabond treated slides (Vector Laboratories, Burlingame, CA) and air dried overnight. The nuclei were then treated with 50% glycerol/2 × SSC for 10 minutes at 90°C and denatured for 10 minutes in 70% formamide/2 × SSC at 70°C. FISH was performed with the ALK probe purchased from Vysis Incorporated (Downers Grove, IL) according to their protocol. Two hundred or more nuclei were scored from each case. The ALK FISH probe generally appears as two distinct signals adjacent to each other (red and green). When there is DNA rearrangement around the ALK region, the probe signal splits. FISH was interpreted as normal without rearrangement when two signals without splits were present, aneuploid without rearrangement when more than two signals without splits were present, and positive for ALK rearrangement when a split signal was present.
Molecular Genetic Studies
Twenty-one specimens from 19 patients were assessed using polymerase chain reaction (PCR) for clonal rearrangements of the T-cell receptor gamma-chain gene as previously described (33). All extracted DNA was checked for amplifiability using a beta-globin PCR.
Statistical Analysis
χ2 and t test analyses were performed, and 95% confidence limits from the binomial theorem were calculated.
RESULTS
Clinical Data
Clinicopathologic features, outcome and presence or absence of ALK abnormalities are summarized in Table 2.
The 40 patients with IMT ranged from newborn to 28 years of age at diagnosis, with a mean age of 8 years. There were 18 males and 22 females (male-to-female ratio 0.8). Thirty tumors (75%) originated in the abdomen, pelvis, or retroperitoneum. Five (13%) arose in the lung, three (7%) arose in the mediastinum and thorax, and two (5%) arose in the extremity. Follow-up information was available for 31 of 40 patients. At last follow-up, 26 patients (84%) had no evidence of disease. Ten patients (32%) experienced one or more recurrences, and five patients (16%) had tumors with histologic evidence of malignant transformation and multiple recurrences. Among the five patients with malignant transformation, two had IMTs with ALK abnormalities and three had aneuploid IMTs without ALK abnormalities (see below).
χ2 analysis did not confirm any association between sex (P =.26), site (P =.74), recurrence (P =.27), or malignant transformation (P =.29) and the presence of ALK abnormalities. The unpaired t test did not show any significant difference in mean age between IMTs with and without ALK abnormalities (P =.32).
Pathologic Findings
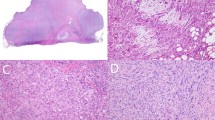
All of the IMTs consisted of spindled myofibroblastic cells accompanied by an inflammatory infiltrate of plasma cells with or without lymphocytes and eosinophils (Fig. 1). The histologic features were identical to previous detailed descriptions (1, 6). Histologic criteria for malignant transformation included increased cellularity, increased numbers of ganglion-like plump polygonal or round cells associated with necrosis, large prominent nucleoli, and variable numbers of mitoses, including atypical mitotic figures (1, 6).

Inflammatory myofibroblastic tumor consists of a combination of spindled myofibroblastic cells and an inflammatory infiltrate with plasma cells.
Immunohistochemical findings were obtained for 47 samples from 40 patients. 16 samples (34%) displayed immunoreactivity for both ALK-1 and p80. One sample (2%) was immunoreactive for ALK-1 and had no immunostaining for p80. 30 samples (64%) were nonreactive for both ALK-1 and p80. All of the cases with immunohistochemical reactivity for ALK-1 and/or p80 displayed cytoplasmic staining without any nuclear staining (Fig. 2).

Immunohistochemistry in inflammatory myofibroblastic tumors. A, ALK-1 cytoplasmic positivity. B, p80 cytoplasmic positivity.
Abnormal karyotypes were obtained in two cases. One IMT from the abdomen had a karyotype of 43–46, x, -y, i(8)(q10),-22, +r, +mar, inc (ALK split by FISH) and were previously published (9). The other, from the lung, had a karyotype of 46, xx, der(22;4;?)(p21;q21;?), der(4)t(2;4;?). Both cases were immunoreactive for ALK-1 and p80, and both had ALK rearrangements by FISH. One case was previously reported (9).
FISH
FISH revealed two distinct adjacent red and green fusion signals in 7 of 22 samples (7 samples from 7 patients). These were interpreted as normal and not rearranged around the ALK region. Four samples had 9 to 87% of cells with more than two fusion signals, indicative of aneuploidy without rearrangement around the ALK region; these four samples were from three patients. One of the aneuploid samples was from a patient with recurrent IMT whose initial tumor had a normal, diploid pattern by FISH. Ten samples had 9 to 64% cells with a split probe signal indicative of DNA rearrangement around the ALK region (Fig. 3); these 10 samples were from nine patients. The split signals were generally seen in large cells. These findings are summarized in Table 3.

Fluorescence in situ hybridization with the ALK probe shows a split of the red and green fusion signals in tumor cell nuclei.
Comparison of immunohistochemistry and FISH results for 19 cases with ALK abnormalities by either method is shown in Table 4. In general, the immunohistochemistry and FISH correlated well, with similar results in 89% of cases. χ2 analysis revealed a strong association between positivity with immunohistochemistry for ALK and p80 and with FISH positivity (P =.00058). Only 2 of 19 cases (10%) did not correlate. One case with rearrangement by FISH had negative immunohistochemistry, and one with no rearrangement by FISH displayed immunoreactivity for ALK-1 and p80. These discrepancies may reflect the technical challenges of ALK immunohistochemistry on referral cases and of FISH on nuclei retrieved from formalin-fixed, paraffin-embedded tissues. None of the aneuploid cases demonstrated immunoreactivity for ALK-1 or p80.
Molecular Genetic Studies
PCR for clonal rearrangement of the T-cell receptor gamma-chain gene was performed in 21 samples from 19 patients. All of the samples yielded a polyclonal smear pattern indicative of absence of clonal rearrangements of the T-cell receptor gene.
Comparison of IMTs with and without ALK Abnormalities
Cases with ALK abnormalities detected by either FISH or immunohistochemistry or both, were compared with cases without evidence of ALK abnormalities. Fourteen of 40 patients (35%) had ALK abnormalities and 26 of 40 (65%) had no detectable abnormalities with the methods used. Comparison of age, sex distribution, site, and outcome are shown in Table 2. Patients with IMT with ALK abnormalities were younger, had a slight male predominance, and a similar anatomic distribution. There was a higher percentage of recurrence in comparison with those without ALK abnormalities. The frequency of malignant transformation was similar in both groups. However, among the five patients with malignant transformation, three had aneuploidy, two of which lacked evidence of ALK rearrangements. The differences in age, sex, site, and recurrence among the 40 patients did not reach statistical significance by χ2 analysis and unpaired t test. The 95% confidence limits demonstrated that IMT occurred more often in the abdomen (observed 75%; limits 58.8 to 87.3%) than in the chest (observed 20%; limits 9.1 to 35.6%) and extremities (observed 5%; limits 0.6 to 16.9%).
Five patients with IMT had aneuploidy, three by FISH and two by flow cytometry (previously published, Ref. 6). A total of 37 samples from 33 patients were assessed for aneuploidy, 21 samples in 19 patients by flow cytometry (previously published, Ref. 6), and 22 samples in 19 patients by FISH; 6 samples in 5 patients were studied with both techniques. These five patients ranged from two to 16 years of age at diagnosis, with a mean of 8 years. There were one male and four females (male-to-female ratio 0.25). The aneuploid IMTs were located in the abdomen, pelvis, or retroperitoneum in two cases (40%), chest in two cases (40%), and extremities in one case (20%). Outcome information was available for four of five patients. One patient had no evidence of disease at last follow-up. Three of the five patients with aneuploid IMT (75%) had recurrences and histologic evidence of malignant transformation. All five aneuploid cases lacked evidence of ALK gene rearrangement by immunohistochemistry or FISH.
DISCUSSION
IMT is classified with intermediate neoplasms in the current World Health Organization Histological Typing of Soft Tissue Tumors (34). It occurs in pulmonary and extrapulmonary sites, is associated with recurrences and systemic symptoms, rarely undergoes malignant transformation, and is composed of differentiated myofibroblastic spindle cell accompanied by numerous plasma cells and/or lymphocytes and eosinophils (1, 3, 4, 5). Some of the persistent uncertainties about IMT are whether it is a benign, indeterminate, or malignant neoplasm, whether it represents a neoplastic or reactive process, what is the significance of multifocal or metastatic disease, and whether histologic-prognostic features can be identified (2, 7, 8). Recent studies have suggested that the combination of ganglion-like cells, aneuploidy, and p53 expression are associated with more aggressive clinical behavior (5, 6, 35).
Several case reports and small series of IMT in the past decade have provided cytogenetic evidence for a clonal origin (4, 9, 10, 11, 12, 13). The clonal abnormalities identified in these reports have varied, but have had in common aberrations involving chromosome 2p21–24. FISH with a probe flanking the ALK gene at 2p23 demonstrated rearrangement associated with ALK expression by immunohistochemistry in three cases (9). A single case of IMT has been reported with a translocation involving 12q15 and an intragenic rearrangement of high mobility group I-C (36). Concurrently with the present report, tropomyosin (TPM)-ALK fusions involving TPM3-ALK and TPM4-ALK oncogenes were reported in two abdominal and one pulmonary IMT (37). In this series, 64% of IMT displayed ALK immunoreactivity but only 27% had ALK fusions by RT-PCR screening for TPM3-ALK and TPM4-ALK. These findings support the concept that IMT is clonal in origin, that ALK rearrangement may be a common theme among this type of neoplasm, and that the ALK fusion genes or ALK expression may be a diagnostically useful molecular marker.
The present study of 47 samples of IMT from 40 patients revealed ALK abnormalities by immunohistochemistry and/or FISH in 35% of the IMTs from 40 patients. Immunoreactivity for ALK-1 and/or p80 was detected in 36% of the 40 patients’ IMTs, and 47% had ALK rearrangement by FISH (P =.00058). 10% lacked correlation of ALK deregulation by FISH versus immunohistochemistry. Comparison of clinicopathologic features and outcome suggested that IMTs with ALK abnormalities may occur at an earlier age, have a male predilection and recur more frequently. However, in this sample of 40 patients, the differences between IMTs with and without ALK abnormalities did not reach statistical significance. Aneuploidy by FISH was not associated with ALK rearrangement, but aneuploid IMT was more frequently associated with recurrence and histologic evidence of malignant transformation. One IMT that was aneuploid by flow cytometry displayed immunoreactivity for ALK-1 and p80, but could not be evaluated by FISH.
It has been suggested that 2p23 contains a gene or regulatory region involved in the pathogenesis of IMT, analogous to the situation for ALCL (9). ALCL comprises a cytologic and morphologic spectrum with many morphologic variants, including a rare sarcomatoid variant that overlaps histologically with IMT (18, 38, 39). Despite morphologic diversity, ALCL has characteristic clinical features and outcome. Most ALCL are derived from T-cells but have a better prognosis than other T-cell lymphomas. ALCL frequently presents with an advanced stage, extranodal involvement, and noncontiguous nodal involvement. There is a bimodal age distribution with peaks in the third and seventh decades. Systemic symptoms, similar to those seen in Hodgkin’s disease and some cases of IMT, are found in 40% of ALCL. Despite a tendency for presentation at high stage and frequent relapses, the prognosis is relatively favorable. Like IMT, ALCL may undergo spontaneous regression (18, 39).
The translocation t(2;5)(p23;p35) has been reported in 15 to 65% of cases of ALCL and variant translocations involving chromosome 2 and other chromosomes have been reported (14, 18, 19, 25, 27, 28). The t(2;5) breakpoint was cloned in 1994 and results in fusion of the NPM gene on chromosome 5q35 to a kinase gene, ALK, on chromosome 2p23 (14). This leads to expression of a fusion protein p80 NPM/ALK. The immunoreactivity for ALK and p80 in ALCL is nuclear and cytoplasmic (19, 29, 40). Although correlation is good between the two antibodies, ALK staining is stronger and easier to interpret. The p80 fusion protein-positive ALCL has been noted to occur in a younger age group with a more favorable outcome and classic lymphohistiocytic or small cell morphologic variants (27, 30). Good correlation has been noted between immunohistochemical staining for antibodies to p80 and ALK, and reverse transcriptase-PCR covering the fusion junction of p80 mRNA (40). Based on studies of ALCL, it appears that a variety of genes are capable of deregulating ALK expression (19, 20, 29, 28).
Rare examples of a sarcomatoid variant of ALCL have been reported (38, 39). The cytogenetic, ALK immunoreactivity, and molecular features of this variant are unknown, but there are overlapping histologic and immunohistochemical features with IMT. The histologic findings of large oval pleomorphic and spindle tumor cells with a storiform pattern and focally myxoid stroma raise the question of a relationship to IMT and other pleomorphic mesenchymal tumors of soft tissue. The absence of T-cell gene rearrangements in the present series of cases provides evidence that IMT is not a T-cell malignant neoplasm or a sarcomatoid variant of ALCL. However, it raises the question of whether there are similar molecular genetic aspects of each tumors that result in morphologic and clinical similarities.
CONCLUSION
This study demonstrates that a significant subset of IMTs are neoplasms with consistent aberrations of chromosome 2p23 and that a second subset lacks evidence of 2p23 abnormalities but is aneuploid and potentially more aggressive. A third group lacks evidence of aneuploidy or ALK abnormalities with the techniques we used. Whether the third subset is characterized by one or more chromosomal aberrations involving regions other than 2p23 is as yet unknown, but the findings suggest a similarity to the ALK-positive and ALK-negative groups of ALCL. There are no distinguishing histologic characteristics to explain the genetic heterogeneity or variations in clinical features. Based on our findings, a significant proportion of IMTs join ALCL as a member of the group of neoplasms with ALK gene involvement. A tentative analogy can be drawn between IMT and ALCL and the EWS family of tumors, in which the EWS gene is involved a group of morphologically similar but clinically and pathologically distinct lesions. Perhaps there is a family of neoplasms, the ALK family, of which ALCL (ALK lymphoma) and IMT with ALK abnormalities are the first recognized members. The recent finding of ALK tyrosine kinase gene expression in neuroblastoma (41) and the identification of specific TPM3-ALK and TPM4-ALK oncogenes in IMT (37) further support this hypothesis. The recognition of specific gene fusion partners of ALK rearrangements in IMT is incomplete, although a recent study has suggested TPM involvement. Whether a specific type of ALK gene fusion has clinical and prognostic significance for IMT remains a matter for further investigation.
References
Coffin CM, Watterson J, Priest JR, Dehner LP . Extrapulmonary inflammatory myofibroblastic tumor (inflammatory pseudotumor). A clinicopathologic and immunohistochemical study of 84 cases. Am J Surg Pathol 1995; 19: 859–872.
Coffin CM, Dehner LP, Meis-Kindblom JM . Inflammatory myofibroblastic tumor, inflammatory fibrosarcoma, and related lesions: an historical review with differential diagnostic considerations. Semin Diag Pathol 1998; 15: 102–110.
Coffin CM, Humphrey PA, Dehner LP . Extrapulmonary inflammatory myofibroblastic tumor: a clinical and pathological survey. Semin Diag Pathol 1998; 15: 85–101.
Hojo H, Newton WA, Hamoudi AB, Qualman SJ, Wakasa H, Suzuki S, et al. Pseudosarcomatous myofibroblastic tumor of the urinary bladder in children: a study of 11 cases with review of the literature. An Intergroup Rhabdomyosarcoma Study. Am J Surg Pathol 1995; 19: 1224–1236.
Biselli R, Ferlini C, Fattorossi A, Boldrini R, Bosman C . Inflammatory myofibroblastic tumor (inflammatory pseudotumor). DNA flow cytometric analysis of nine pediatric cases. Cancer 1996; 77: 778–784.
Hussong JW, Brown M, Perkins SL, Dehner LP, Coffin CM . Comparison of DNA ploidy, histologic, and immunohistochemical findings with clinical outcome in inflammatory myofibroblastic tumors. Mod Pathol 1999; 12: 279–286.
Chan JKC . Inflammatory pseudotumor: a family of lesions of diverse nature and etiologies. Adv Anat Pathol 1996; 3: 156–171.
Meis JM, Enzinger FM . Inflammatory fibrosarcoma of the mesentery and retroperitoneum. A tumor closely simulating inflammatory pseudotumor. Am J Surg Pathol 1991; 15: 1146–1156.
Griffin CA, Hawkins AL, Dvorak C, Henkle C, Ellingham T, Perlman EJ . Recurrent involvement of 2p23 in inflammatory myofibroblastic tumors. Cancer Res 1999; 59: 2776–2780.
Sciot R, Dal Cin P, Fletcher CDM, Hernandez JM, Garcia JL, Samson I, et al. Inflammatory myofibroblastic tumor of bone. Report of two cases with evidence of clonal chromosomal changes. Am J Surg Pathol 1997; 21: 1166–1172.
Snyder CS, Dell ’Aquila M, Haghighi P, Baergen RN, Suh YK, Yi ES . Clonal changes in inflammatory pseudotumor of the lung. A case report. Cancer 1995; 76: 1545–1549.
Su LD, Atyde-Perez A, Sheldon S, Fletcher JA, Weiss SW . Inflammatory myofibroblastic tumor: cytogenetic evidence supporting clonal origin. Mod Pathol 1998; 11: 364–368.
Treissman SP, Gillis A, Lee CLY, Giacomantonio M, Resch L . Omental-mesenteric inflammatory pseudotumor. Cytogenetic demonstration of genetic changes and monoclonality in one tumor. Cancer 1994; 73: 1433–1437.
Morris SE, Kirstein MN, Valentine MB, Dittmer KG, Shapiro DN, Saltman DL, et al. Fusion of a kinase gene, ALK, to a molecular protein gene, NPM, in non-Hodgkin’s lymphoma. Science 1994; 263: 1281–12814.
Benharroch D, Meguerian-Bedoyan Z, Lamant L, Amin C, Brugieres L, Terrier-Lacombe MJ, et al. ALK-positive lymphoma: a single disease with a broad spectrum of morphology. Blood 1998; 91: 2076–2084.
Falini B, Bignera B, Fizzotti M, Pulford K, Pileri SA, Delsol G, et al. ALK expression defines a distinct group of T-null lymphomas (“ALK lymphomas”) with a wide morphological spectrum. Am J Pathol 1998; 153: 875–886.
Herbst H, Anagnostopoulos J, Heinze B, Dürkop J, Hummel M, Stein H . ALK gene products in anaplastic large cell lymphomas and Hodgkin’s disease. Blood 1995; 86: 1694–1700.
Kadin ME . Anaplastic large cell lymphoma and its morphological variants. Cancer Surv 1997; 30: 77–86.
Lamant L, Meggetto F, Saati TA, Brugières L, Bressac de Paillerets B, et al. High incidence of the t(2;5)(p23;q35) translocation in anaplastic large cell lymphoma and its lack of detection in Hodgkin’s Disease. Comparison of cytogenetic analysis, reverse transcriptase-polymerase chain reaction, and p-80 immunostaining. Blood 1996; 87: 284–291.
Yee HT, Ponzoni M, Merson A, Goldstein M, Scarpa A, Chilosi M, et al. Molecular characterization of the t(2;5)(p23;q35) translocation in anaplastic large cell lymphoma (Ki-1) and Hodgkin’s disease. Blood 1996; 87: 1081–1088.
Colleoni GW, Bridge JA, Garicochea B, Liu J, Filippa DA, Ladanyi M . ATIC-ALK. A novel variant ALK gene fusion in anaplastic large cell lymphoma resulting from the recurrent cryptic chromosomal inversion, inv(2)(p23q35). Am J Pathol 2000; 156: 781–789.
Hernandez L, Pinyol M, Hernandez S, Bea S, Pulford K, Rosenwald A, et al. TRK-fused gene (TFG) is a new partner of ALK in anaplastic large cell lymphoma producing two structurally different TFG-ALK translocations. Blood 1999; 94: 3265–3268.
Lamant L, Dastugue N, Pulford K, Delsol G, Mariame B . A new fusion gene TPM3-ALK in anaplastic large cell lymphoma created by a (1;2)(q25;p23) translation. Blood 1999; 93: 3088–3095.
Ma Z, Cools J, Marynen P, Cui X, Siebert R, Gesk S, et al. Inv(2)(p23q35) in anaplastic large-cell lymphoma induces constitutive anaplastic lymphoma kinase (ALK) tyrosine kinase activation by fusion to ATIC, an enzyme involved in purine nucleotide biosynthesis. Blood 2000; 95: 2144–2149.
Touriol C, Greenland C, Lamant L, Pulford K . Bernard F, Rousset T, et al. Further demonstration of the diversity of chromosomal changes involving 2p23 in ALK-positive lymphoma: 2 cases expressing ALK kinase fused to CLTCL (clathrin chain polypeptide-like). Blood 2000; 95: 3204–3207.
Trinei M, Lanfrancone L, Campo E, Pulford K, Mason DY, Pelicci PG, et al. A new variant anaplastic lymphoma kinase (ALK)-fusion protein (ATIC-ALK) in a case of ALK-positive anaplastic large cell lymphoma. Cancer Res 2000; 60: 793–798.
Nakamura S, Shiota M, Nakagawa A, Yatabe Y, Kojima M, Motoori T, et al. Anaplastic large cell lymphoma: a distinct molecular pathologic entity. A reappraisal with special reference to p80NPM/ALK expression. Am J Surg Pathol 1997; 21: 1420–1432.
Pittaluga S, Wlodarska I, Pulford K, Campo E, Morris SW, Van den Berghe H, et al. Short communication. The monoclonal antibody ALK-1 identifies a distinct morphological subtype of anaplastic large cell lymphoma associated with 2p23/ALK rearrangements. Am J Pathol 1997; 151: 343–351.
Pulford K, Lamant L, Morris SW, Butler LH, Wood KM, Stroud D, et al. Detection of anaplastic lymphoma kinase (ALK) and nucleolar protein nucleophosmin (NPM)-ALK proteins in normal and neoplastic cells with the monoclonal antibody ALK1. Blood 1997; 89: 1394–1404.
Shiota M, Nakamura S, Ichinohasama R, Masafumi A, Tadaatsu A, Takeshita M, et al. Anaplastic large cell lymphomas expressing the novel chimeric protein p80NPM/ALK: a distinct clinicopathologic entity. Blood 1995; 86: 1954–1960.
Carey MJ, Medeiros LJ, Roepke JE, Kjeldsberg CR, Elenitoba-Johnson KSJ . Primary anaplastic large cell lymphoma of the small intestine. Am J Clin Pathol 1999; 112: 696–701.
Visakorpi T, Hyytinen E, Kallioniemi A, Isola J, Kallioniemi OP . Sensitive detection of chromosome copy number aberrations in prostate cancer by fluorescence in situ hybridization. Am J Pathol 1994; 145: 624–630.
McCarthy KP, Sloane JP, Kabarowski JH, Matutes E, Wiedemann LM . A simplified method of detection of clonal rearrangements to the T-cell receptor-chain gene. Diag Mol Pathol 1992; 1: 173–179.
Weiss SW . Histologic Typing of Soft Tissue Tumors, 2nd Ed. New York: Springer-Verlag; 1994. p. 48.
Biselli R, Boldrini R, Ferlini C, Boglin C, Inserra A, Bosman C . Myofibroblastic tumors: neoplasias with divergent behavior. Ultrastructural and flow cytometric analysis. Pathol Res Pract 1999; 195: 619–632.
Kazmierczak B, Dal Cin P, Sciot R, Van den Berghe H, Bullerdiek J . Inflammatory myofibroblastic tumor with HMGIC rearrangement. Cancer Genet Cytogenet 1999; 112: 156–160.
Lawrence B, Perez-Atayde A, Hibbard MK, Rubin BP, Dal Cin P, Pinkus JL, et al. TPM3-ALK and TPM4-ALK oncogenes in inflammatory myofibroblastic tumors. Am J Pathol 2000; 157: 377–384.
Chan JKC, Buchanan R, Fletcher CDM . Sarcomatoid variant of anaplastic large-cell Ki-1 lymphoma. Am J Surg Pathol 1990; 14: 983–988.
Kashiwabara K, Nakamura H, Kusama J, Kishi K, Yagyu H, Takagi S, et al. Ki-1 (CD30)-positive anaplastic large cell lymphoma, sarcomatoid variant accompanied by spontaneously regressing lymphadenopathy. Intern Med 1999; 38: 49–53.
Shiota M, Fujimoto J, Takenaga M, Satoh H, Ichinohasama R, Abe M, et al. Diagnosis of t(2;5)(p23;q35)-associated Ki-1 lymphoma with immunohistochemistry. Blood 1994; 84: 3648–3652.
Lamant L, Pulford K, Bischof D, Morris SW, Mason DY, Delsol G, et al. Expression of the ALK tyrosine kinase gene in neuroblastoma. Am J Pathol 2000; 156: 1711–1721.
Acknowledgements
The authors thank Michelle González for typing the manuscript, Sheryl Tripp of ARUP Laboratories and The ARUP Institute for technical assistance, and Ed Ashwood, M.D., Ph.D., of the University of Utah Department of Pathology, for statistical analysis.
Author information
Authors and Affiliations
Rights and permissions
About this article
Cite this article
Coffin, C., Patel, A., Perkins, S. et al. ALK1 and p80 Expression and Chromosomal Rearrangements Involving 2p23 in Inflammatory Myofibroblastic Tumor. Mod Pathol 14, 569–576 (2001). https://doi.org/10.1038/modpathol.3880352
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.3880352
Keywords
This article is cited by
-
Intraocular myofibroblastoma tumour of the ciliary body: a case report and literature review
BMC Ophthalmology (2022)
-
S-100 Immuno-Positive Low Grade Myofibroblastic Sarcoma of Nasal Cavity: A Rare Case Presentation and Review of Literature
Indian Journal of Otolaryngology and Head & Neck Surgery (2022)
-
Mutations in ALK signaling pathways conferring resistance to ALK inhibitor treatment lead to collateral vulnerabilities in neuroblastoma cells
Molecular Cancer (2022)
