
Abstract
We report an 18-month-old Japanese girl with purely epithelioid leiomyosarcoma presenting as a huge intraabdominal mass. The patient had been well from birth and had shown no signs of immunodeficiency. She was negative for human immunodeficiency virus. Blood examination revealed elevated serum neuron specific enolase (NSE). Histologically, the tumor was comprised of solid growths of round or polygonal cells with vesicular nuclei and often vacuolated cytoplasm rich in glycogen. The tumor cells were positive for vimentin, NSE, and MIC2, and were negative for desmin and neurofilament. The age, clinical presentation, and histologic findings mostly favored Ewing’s sarcoma/primitive neuroectodermal tumor. Silver stain, however, demonstrated well-developed reticulin fibers often outlining individual tumor cells. An expanded panel of immunostains showed that the tumor cells were intensely positive for smooth muscle actin, and ultrastructural study revealed abundant fine cytoplasmic filaments with focal subsarcolemmal densities, various amounts of glycogen, and irregularly arranged, thick basal lamina. The diagnosis of epithelioid leiomyosarcoma was made. Following reduction in tumor size by chemotherapy, the serum NSE level was normalized. From the surgical finding, the primary site was presumed to be the urachus or the urinary bladder dome. Although extremely rare, epithelioid leiomyosarcoma should be added in the list of differential diagnoses of pediatric “round cell tumors.”
Similar content being viewed by others
INTRODUCTION
Leiomyosarcoma (LMS) has become the second leading malignancy of children with human immunodeficiency virus (HIV) infection or other immunodeficiency diseases, and is often associated with Epstein-Barr virus (EBV) infection (1, 2). However, LMS is rare in non-immunocompromised children and accounts for only 2 to 4% of childhood soft tissue sarcomas (3, 4). Epithelioid LMS, predominantly composed of rounded or polygonal epithelioid cells, most often occurs in the gastrointestinal tract of adults more than 20 years of age (5). Epithelioid LMS is extremely rare in patients less than 10 years of age. To the best of our knowledge, there has been no case of infantile epithelioid LMS in the literature. We describe a rare example of purely epithelioid LMS occurring in a non-immunocompromised 18-month-old girl, and refer to a differential diagnosis of round cell tumors in early pediatric age group.
Clinical Summary
An 18-month-old Japanese girl was referred to our center because of marked abdominal distension. No family or personal histories were contributory. The patient’s serum level of neuron specific enolase (NSE) was elevated to 65ng/mL (normal, <10 ng/mL). Anti-HIV antibodies were negative and the other laboratory data showed no remarkable abnormalities. Imaging studies disclosed a large intraabdominal tumor that compressed the intestine and the urinary bladder, but the primary site could not be determined (Fig. 1). Tumor biopsy was performed and the patient underwent adjuvant chemotherapy (intergroup rhabdomyosarcoma study regimen 33). The tumor has responded well to the chemotherapy and the volume decreased to less than half the original size. The serum NSE level was normalized within one month. Surgical intervention was performed 2 mo after the onset. Most parts of the tumor were sharply demarcated from the surrounding organs, but boundaries between the urinary bladder dome and the periumbilical soft tissue were indistinct. The tumor was fed by a single artery from the left internal iliac artery. The median umbilical ligament was not found. Although the primary site of the present tumor was difficult to determine, the growth pattern and the blood supply favored the origin from the urachus or the urinary bladder dome. A complete removal of the primary tumor attached by the urinary bladder dome was achieved; however, a small implantation on serosa of the ileum was found. The patient has been receiving adjuvant chemotherapy for 7 mo until now. No distant metastases have been found during the clinical course.

Abdominal magnetic resonance imaging demonstrating a large tumor (T) extending from the pelvis up to the abdominal wall (UB, urinary bladder).
MATERIALS AND METHODS
For light microscopic examination, tissue samples were fixed in 10% formalin overnight, dehydrated, embedded in paraffin, and cut into 3-μm sections. Hematoxylin and eosin stain (H & E), periodic acid-Schiff reaction, with or without diastase predigestion, and silver stain were performed in all sections. Immunostains using antibodies listed in Table 1 were performed in selected sections. In situ hybridization for EBV-encoded small RNAs was performed according to the manufacturer’s instruction. For ultrastructural observation, 1 × 1-mm tissue blocks were fixed in 2% glutaraldehyde, postfixed in osmium-tetrachloride, dehydrated, embedded in epon, cut into ultrathin sections, double-stained with uranylacetate and lead-citrate, and observed by electron microscopy (JEM 1010; JEOL, Akishima, Japan). Snap-frozen samples were studied for EWS/FLI1 or EWS/ERG chimeric transcripts that are highly specific for Ewing’s sarcoma/primitive neuroectodermal tumor (ES/PNET), as described previously (6, 7).
Pathologic Findings
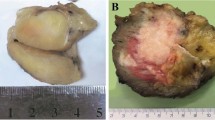
The biopsied sample measured 15 × 12 × 10 mm, and the cut surface showed an elastic-hard, gray appearance. Microscopically, the tumor comprised of relatively large polygonal cells with round or oval nuclei and often vacuolated pale cytoplasm. The tumor cells were arranged in a solid or epithelioid fashion (Fig. 2A). Arrangement in small cords was occasionally encountered. No spindle cell components were observed. Nuclear pleomorphism was not prominent, but mitoses were frequent (6 to 8 per 10 high power fields [HPFs]). Numerous periodic acid-Schiff-positive granules were found in the cytoplasm and were digested by diastase. The tumor cells were diffusely positive for vimentin and MIC2 (Fig. 2B), focally positive for NSE and cytokeratin, and were completely negative for desmin, myoglobin, neurofilament, epithelial membrane antigen (EMA), leukocyte common antigen (LCA), S-100, and c-kit. However, silver stain clearly demonstrated well-developed reticulin fibers often outlining individual tumor cells (Fig. 2C). Additional immunostaining revealed the tumor cells to be diffusely positive for smooth muscle actin (Fig. 2D), and focally positive for muscle actin, calponin, and caldesmon. In situ hybridization for EBV-encoded small RNAs was negative. Ultrastructural studies demonstrated abundant fine cytoplasmic filaments with focal subsarcolemmal densities, a considerable amount of glycogen, irregularly arranged basal lamina, and occasional intercellular attachments (Fig. 3, A-B). Final diagnosis was epithelioid LMS.

A, histology of the tumor showing solid and epithelioid growth of polygonal cells. Note the vesicular nuclei and often vacuolated cytoplasm. Mitosis was occasionally seen (hematoxylin and eosin stain; original magnification, 100 ×; inset, 200 ×). B, positive immunostaining for MIC2 (hematoxylin counter-stain; original magnification, 200 ×). C, tumor cells demarcated by reticulin fibers in single cells (silver stain; original magnification, 100 ×). D, positive immunostain for smooth muscle actin (hematoxylin counterstain; original magnification, 200 ×).

Ultrastructure: Tumor cells containing intermediate filaments, thin filaments with subplasmalemmal dense bodies (thin arrows), and glycogen particles (G). Note a formation of thick and irregular basement membranes (thick arrows). Original magnification: A, 6000 ×; B, 12,000 ×.
The surgically removed tumor measured 15.5 × 12.5 × 5.3 cm, weighed 510 g, and was well demarcated. Cut surfaces were yellowish, edematous, and partly hemorrhagic. Histologic examination revealed small nests or cords of tumor cells with an extensively myxoid background in which were scattered hemosiderin-laden histiocytes. The tumor cells showed a similar morphology to the biopsy sample. No spindle cell components were observed. The tumor merged with outer muscular layer of the urinary bladder dome and was completely separated from the periumbilical soft tissue by fibrous capsule. The implantation on subserosa of the ileum showed a similar histopathology to the primary tumor, but the proportion of the viable tumor cells was higher than the primary tumor.
Molecular Findings
No chimeric transcripts of EWS/FLI1 or EWS/ERG were detected both in the biopsy sample and the surgically obtained tissue.
DISCUSSION
Histopathology of the present tumor was similar to “round cell tumor” and the vacuolated cytoplasm was rich in glycogen. Furthermore, the tumor cells were positive for vimentin, NSE, and MIC2, and were negative for desmin, myoglobin, LCA, and neurofilament protein. The above-mentioned histopathology, the patient’s age (1.5 years), the primary site (abdominal cavity), and the raised serum NSE level (8), mostly favored a diagnosis of ES/PNET group tumors. Silver stain, however, demonstrated well-developed reticulin fibers often surrounding individual tumor cells that were unlikely for ES/PNET group tumors (9). This finding prompted us to perform immunostains for smooth muscle markers and ultrastructural studies that confirmed the diagnosis of epithelioid LMS.
MIC2 has been known as a sensitive marker for ES/PNET group tumors, but recent studies disclosed that most other tumors occasionally show distinct MIC2 staining (10, 11). MIC2 positivity in LMS has not been well established. Ambros et al. (10) reported that none of the 6 LMS was positive for MIC2 upon examination using formalin-fixed, paraffin-embedded tissues. Fellinger et al. (11) showed that six of 11 LMS were positive for MIC2 upon studies using frozen sections. Negative results of myogenic markers are not rare in LMS, and desmin is more often negative than muscle actin and smooth muscle actin (12). Focal positive results of cytokeratin are consistent with LMS (12).
The elevated serum NSE level and the positive immunostaining for NSE in tumor cells made the initial diagnosis more confusing. These findings, however, have been described in LMS cases (13, 14), and are understandable because expression of NSE has been reported in various tissues, including smooth muscle cells and smooth muscle tumors (13, 15). Moreover, enolase is one of the glycolytic enzymes. Induction of this enzyme may occur in proliferating tumor cells under hypoxic condition.
The most frequent locations of LMS in non-immunocompromised children are the gastrointestinal tract and more than half the cases occur in the third quinquennium (5). LMS in non-immunocompromised children less than 5 years of age are quite rare. A literature review showed approximately 20 cases with no example of epithelioid LMS (3, 5, 12, 16, 17, 18). Also, epithelioid LMS has never been reported in an immunocompromised child. Intraabdominal LMS, including LMS of the retroperitoneum and the urinary bladder, is common in adults but is extremely rare in early childhood. Other tumors, such as rhabdomyosarcoma, ES/PNET, neuroblastoma, desmoplastic small cell tumor, and malignant lymphoma are overwhelmingly common as intraabdominal malignancies in early childhood.
Although the present case responded well to adjuvant chemotherapy, LMS generally does not show good response to chemotherapy as compared with other sarcomas, such as ES/PNET. The first treatment of choice is complete surgical excision that correlates best with a favorable outcome (3). Epithelioid LMS should be added in the list of differential diagnoses of pediatric “round cell tumors” to avoid possible misdiagnosis.
References
Granovsky MO, Mueller BU, Nicholson HS, Rosenberg PS, Rabkin CS . Cancer in human immunodeficiency virus-infected children. J Clin Oncol 1998; 16: 1729–1735.
Tulbah A, Al-Dayel F, Fawaz I, Rosai J . Epstein-Barr virus-associated leiomyosarcoma of the thyroid in a child with congenital immunodeficiency: a case report. Am J Surg Pathol 1999; 23: 473–476.
Angel CA, Gant LL, Parham DM, Rao BN, Douglass EC, Lobe T, et al. Leiomyosarcomas in children: clinicopathologic characteristics. Pediatr Surg Int 1992; 7: 116–120.
Harms D . Soft tissue sarcomas in the Kiel Pediatric Tumor Registry. Curr Top Pathol 1995; 89: 31–45.
Lack EE . Leiomyosarcomas in childhood: a clinical and pathologic study of 10 cases. Pediatr Pathol 1986; 6: 181–197.
Delattre O, Zucman J, Melot T, Garau XS, Zucker JM, Lenoir GM, et al. The Ewing family of tumors–A subgroup of small-round-cell tumors defined by specific chimeric transcripts. N Engl J Med 1994; 31: 294–299.
Sorensen PHB, Lessnick SL, Lopez-Terrada D, Liu XF, Triche TJ, Denny CT . A second Ewing’s sarcoma translocation, t(21;22), fuses the EWS gene to another ETS-family transcription factor, ERG. Nat Genet 1994; 6: 146–151.
Fizazi K, Le Cesne A, Dohollou N, Affaied S, Spielmann M, Le Chevalier T . Serum neuron-specific enolase (NSE) as a tumour marker for the Ewing’s sarcoma family of tumours. Eur J Cancer 1996; 32A: 1823–1824.
Angervall L, Enzinger FM . Extraskeletal neoplasm resembling Ewing’s sarcoma. Cancer 1975; 36: 240–251.
Ambros IM, Ambros PF, Strehl S, Kovar H, Gadner H, Salzer-Kuntschik M . MIC2 is a specific marker for Ewing’s sarcoma and peripheral primitive neuroectodermal tumor. Cancer 1991; 67: 1886–1893.
Fellinger EJ, Garin-Chesa P, Triche TJ, Huvos AG, Rettig WJ . Immunohistochemical analysis of Ewing’s sarcoma cell surface antigen p30/32MIC2. Am J Pathol 1991; 139: 317–325.
de Saint Aubain Somerhausen N, Fletcher CDM . Leiomyosarcoma of soft tissue in children. Clinicopathologic analysis of 20 cases. Am J Surg Pathol 1999; 23: 755–763.
Usui A, Kato K, Abe T, Hachisuka K, Yamaguchi A . Creatine kinase BB and neuron-specific gamma-enolase are enhanced in leiomyosarcoma. Clin Chim Acta 1988; 175: 109–111.
Dobashi Y, Nasuno S, Noguchi T, Kameya T . Neuron-specific enolase-producing epithelioid leiomyosarcoma with gross vascular invasion and intracardiac involvement. Arch Pathol Lab Med 1998; 122: 1116–1119.
Kato K, Ishiguri Y, Suzuki F, Ito A, Semba R . Distribution of nervous system-specific forms of enolase in peripheral tissues. Brain Res 1982; 237: 441–448.
Hwang ES, Gerald W, Wollner N, Meyers P, La Quaglia MP . Leiomyosarcoma in childhood and adolescence. Ann Surg Oncol 1997; 4: 223–227.
Jimenez CL, Carpenter BF, Robb I . Leiomyosarcoma. Ultrastruct Pathol 1987; 11: 765–769.
Swanson PE, Wick MR, Dehner LP . Leiomyosarcoma of somatic soft tissues in childhood: an immunohistochemical analysis of six cases with ultrastructural correlation. Hum Pathol 1991; 22: 569–577.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Kato, K., Arai, K., Tanaka, Y. et al. Epithelioid Leiomyosarcoma in a Non-Immunocompromised Infant: Additional Differential Diagnosis of Pediatric “Round Cell Tumors”. Mod Pathol 13, 1156–1160 (2000). https://doi.org/10.1038/modpathol.3880213
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.3880213
Keywords
This article is cited by
-
Structure and function of ETAA16: a novel cell surface antigen in Ewing’s tumours
Cancer Immunology, Immunotherapy (2006)
