
Abstract
Primary angiosarcoma of the spleen is a rare neoplasm that has not been well characterized. We describe the clinical, morphologic, and immunophenotypic findings of 28 cases of primary splenic angiosarcoma, including one case that shares features of lymphangioma/lymphangiosarcoma. The patients included 16 men and 12 women, aged 29 to 85 years, with a mean of 59 years and median of 63 years. The majority of patients (75%) complained of abdominal pain, and 25% presented with splenic rupture. The most common physical finding was splenomegaly (71%). Seventeen of 21 patients were reported to have anemia. Macroscopic examination showed splenomegaly in 85% cases. Sectioning revealed discrete lesions in 88% of cases, ranging from well-circumscribed firm nodules to poorly delineated foci of necrosis and hemorrhage associated with cystic spaces. Microscopically, the tumors were heterogenous; however, all cases demonstrated at least a focal vasoformative component lined by atypical endothelial cells. Solid sarcomatous, papillary, and epithelioid growth patterns were observed. The solid sarcomatous component resembled fibrosarcoma in two cases and malignant fibrous-histiocytoma in one case. Hemorrhage, necrosis, hemosiderin, extramedullary hematopoiesis, and intracytoplasmic hyaline globules were frequently identified. A panel of immunohistochemical studies revealed that the majority of tumors were immunoreactive for at least two markers of vascular differentiation (CD34, FVIIIRAg, VEGFR3, and CD31) and at least one marker of histiocytic differentiation (CD68 and/or lysozyme). Metastases developed in 100% of patients during the course of their disease. Twenty-six patients died of disease despite aggressive therapy, whereas only two patients are alive at last follow-up, one with disease at 8 years and the other without disease at 10 years. In conclusion, primary splenic angiosarcoma is an extremely aggressive neoplasm that is almost universally fatal. The majority of splenic angiosarcomas coexpress histiocytic and endothelial markers by immunohistochemical analysis, which suggest that some tumors may originate from splenic lining cells.
Similar content being viewed by others


INTRODUCTION
Primary malignant vascular neoplasms of the spleen are rare and carry a dismal prognosis regardless of the treatment regimen (1, 2, 3, 4, 5). Although all cases demonstrate at least a focal vasoformative component, malignant splenic vascular tumors may be mistaken for benign vascular or malignant non-vascular tumors because of their frequently highly variable histology. The pathologic features and a comprehensive immunohistochemical profile have not been well addressed in the literature to date.
We describe the clinical, pathologic, and immunophenotypic features of 28 splenic angiosarcomas and analyze this data as it relates to disease course and patient outcome. We also compare the immunohistochemical features of these tumors to three benign primary splenic tumor subtypes: lymphangioma, hemangioma, and littoral cell angioma.
MATERIALS AND METHODS
Fifty-three cases of angiosarcoma involving the spleen were selected from a review of 376 splenectomy specimens retrieved from the archives of the Hematopathology Tumor Registry at the Armed Forces Institute of Pathology (AFIP), Washington, DC, between 1970 and 1998. Twenty-five of the 53 cases of angiosarcoma were excluded from further study because they did not fulfill our inclusion criteria: primary origin in the spleen, sufficient material for immunohistochemical analysis, and adequate clinical information. The remaining 28 cases represented approximately 7.4% of all benign or malignant primary splenic tumors (376) seen in consultation during this time. Primary origin in the spleen was defined as no metastasis at the time of presentation as determined by sophisticated radiologic procedures and/or surgical staging (18/28 cases), or overwhelming tumor burden in the spleen in cases with concomitant metastasis (10/28 cases). The spleen was virtually replaced by the malignancy in the majority of the latter cases. Twenty-three cases were obtained from civilian sources and five cases from military hospitals and Veterans Administration Medical Centers.
Materials within the files of the AFIP were supplemented by a review of the patient demographics, signs and symptoms at presentation, duration of symptoms before presentation, medical history, radiographic studies performed, laboratory test results, surgical pathology and operative reports, cancer registry records, and by written questionnaires or oral communication with the treating physician(s). Follow-up data included information regarding environmental exposures, occupational history, the specific type(s) and length of treatment methods used, and the current status of the disease and patient. This clinical investigation was conducted in accordance and compliance with all statutes, directives, and guidelines of the Code of Federal Regulations, Title 45, Part 46, and the Department of Defense Directive 3216.2 relating to human subjects in research.
Hematoxylin and eosin (H&E) and periodic acid-Schiff stained slides for all cases were reviewed to confirm that established light microscopic histopathologic criteria for angiosarcoma were fulfilled (3). Criteria include the presence of a vasoformative component and cytologic features of malignancy, including cellular atypia and mitotic activity. Five of our cases were previously reported (3). A number of histomorphologic characteristics were tabulated for each case, including histologic growth pattern(s), presence or absence of necrosis, hemorrhage, hemosiderin, hyaline globules, hemophagocytosis, calcification, thrombosis, extramedullary hematopoiesis, and fibrosis. Nuclear atypia was separated into three grades (mild, moderate, and severe) with benign endothelial cells as a reference. Mild atypia referred to nuclei with minimal pleomorphism and hyperchromasia, severe atypia referred to nuclei showing frank anaplasia with large irregular nuclei with hyperchromasia, and moderate atypia referred to those nuclei that fell between the two extremes. Mitotic figures were counted on an Olympus BH-2 microscope at 40 ×. The areas with the highest number of mitoses were chosen for counting.
Immunophenotypic studies were performed on a single block by the standardized avidin-biotin method of Hsu et al. (6) using 4-μm thick sections on all formalin-fixed, paraffin-embedded cases of splenic angiosarcoma, 10 cases of littoral cell angioma, and nine cases each of splenic hemangioma and splenic lymphangioma. Table 1 indexes the immunohistochemical antibody panel chosen. When required, proteolytic antigen retrieval was performed with predigestion for 3 min with 0.05% Protease VIII (Sigma Chemical Co., St. Louis, MO) in a 0.1-m phosphate buffer at a pH of 7.8 at 37° C (7). Antigen enhancement (recovery) was performed, as required, using formalin-fixed, paraffin-embedded tissue treated with a buffered citric acid solution and heated for 20 min in a calibrated microwave oven (8). Following this, the sections were allowed to cool at room temperature in a citric acid buffer solution for 45 min before the procedure was continued. Standard positive controls were used throughout, with serum used as the negative control.
Ten cases of littoral cell angioma and nine cases each of splenic lymphangioma and splenic hemangioma were selected for comparative immunohistochemical analysis.
Categoric variables were analyzed using χ2 tests to compare observed and expected frequency distributions. Fisher’s exact test was used as a substitute for the χ2 test when expected frequencies were fewer than five. Means differences between groups were made with unpaired t tests. Pearson’s correlation coefficient was calculated to evaluate the strength of the association between continuous variables. The Cox proportional hazards model was used to evaluate the association between histologic features and survival. Confidence intervals of 95% were generated for all positive findings. The alpha level was set at P < .05. All analyses were conducted using the computer program, Statistical Package for the Social Sciences (SPSS) 8.0 for PC (Chicago, IL).
RESULTS
Clinical Features
The patients included 16 men and 12 women, aged 29 to 85 years, with a mean age of 59 years at presentation (median, 63 years). The mean age at presentation for women was statistically significantly older (68 years) than men (53 years) (P = .019). The patients experienced abdominal pain (n = 21), weakness or fatigue (n = 11), fever (n = 6), chest pain (n = 3), and/or weight loss (n = 3). Additional symptoms included one patient each with shortness of breath, bleeding ulcers of the gastrointestinal tract, hemoptysis, right flank pain, and anorexia. Two cases were discovered incidentally, one during a work-up for thrombocytopenia and another during staging for malignant lymphoma. The duration of symptoms ranged from less than 1 mo to 7 mo with an average of approximately 2 mo. The length of symptoms did not correlate statistically with overall patient outcome. Physical examination yielded splenomegaly in 20 patients and hepatomegaly in four patients. Seven patients were found to have splenic rupture. Rupture was not associated with outcome.
Laboratory Analysis
Laboratory data was available for 21 patients. The most common laboratory finding was anemia (n = 13). Four patients had leukocytosis and four patients had thrombocytopenia. Additional laboratory abnormalities included one patient each with increased α-1 globulins, elevated liver enzymes, increased lactate dehydrogenase, and metabolic acidosis.
Roentgenographic Findings
Roentgenographic results were reported in 12 patients. Computed tomography (CT) and intravenous pyelogram (IVP) were the most frequently used modalities. In six patients, CT scans demonstrated one to several splenic masses, variably described as hypodense, in-homogeneous, and heterogeneous. In three patients, IVPs showed a left suprarenal mass that was associated with displacement of the kidney in two patients. Additional studies included an ultrasound (showing a massively enlarged spleen) and two liver-spleen scans (yielding multiple nodules or filling defects).
Treatment and Patient Outcome
All patients were treated with splenectomy, with adjuvant therapy employed in only a minority of the patients (Table 2). Five patients received adjuvant radiation therapy alone, four received chemotherapy alone, and two were treated with combination radiation and chemotherapy.
Follow-up information was available for 27 of the 28 patients (Table 2). Only two patients were alive at last follow-up, one with evidence of disease at 8 years (case 19), and one without evidence of disease at 10 years (case 9). The patient who was alive with disease received combination chemotherapy and radiation therapy, whereas the patient without evidence of disease was treated by splenectomy alone.
The remaining 25 patients died with disseminated tumor (mean, 11 mo; median, 5 mo) within 29 mo from initial diagnosis, irrespective of the type of treatment rendered. The majority of patients (n = 16) died within 9 mo (Table 2). There was no statistically significant correlation between survival and treatment rendered (P = .167 and .910, for chemotherapy and radiation, respectively). Most patients developed metastases to the liver (n = 24), followed in decreasing order by metastatic disease to the lung (n = 21), lymph nodes (n = 15), bone (n = 12), soft tissue (n = 7), gastrointestinal tract (n = 6), brain (n = 6), and adrenal glands (n = 4). Additional sites included one patient each with metastasis to the abdominal wall, heart, and pancreas. The majority of patients demonstrated multifocal metastatic deposits in many different organ sites (n = 24).
Pathology
Macroscopic features
The splenic weight ranged from 110 to 3200 g, with a mean of 1073 g. There was splenomegaly in 23 of 27 (85%) of cases, defined as a weight greater than or equal to 250 g (Table 3). Furthermore, 15 of these cases qualified as massive splenomegaly defined as a weight greater than or equal to 1000 g. The cut surface was described as nodular, necrotic, spongy, and hemorrhagic. Discrete lesions were noted in 23 cases. The tumors ranged from well-circumscribed, firm nodules to poorly delimited necrotic and hemorrhagic masses with cystic spaces. A dominant mass was noted in 10 cases, and ranged in size from 1.5 to 18 cm (mean, 8.5 cm), but the overall tumor size did not correlate to clinical outcome (P = .590).
Histologic features
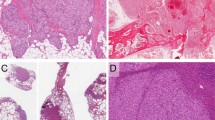
The tumor characteristics were remarkably variable both within and between cases. At low-power magnification, most tumors had a nodular, homogenous appearance (n = 20), adjacent to uninvolved splenic tissue in 22 cases. In the majority of cases, the tumors were infiltrative on higher power magnification. Only four tumors appeared well circumscribed and three tumors spared the splenic white pulp (Fig. 1), a previously unreported finding. A vasoformative component was seen in all cases, and in most cases was the dominant pattern. The tumors showed mild (n = 6), moderate (n = 15), or severe (n = 7) cytologic atypia. In general, tumors with mild atypia showed a dominant vasoformative pattern and were sometimes difficult to distinguish from benign vascular tumors. Tumors with severe atypia frequently were limited in their vasoformative capacity. Bizarre tumor giant cells were observed only in cases with moderate (n = 6) or severe (n = 6) cytologic atypia, a finding that was statistically significant (P = .002).

Angiosarcoma replaces the splenic red pulp and spares the splenic white pulp.
The vascular spaces were variably “slit-like” (Fig. 2A) to “honeycomb-like” (Fig. 2B) in 23 cases, capillary-like in six cases (Fig. 3A), and cavernous in five cases. Two of the cases with slit-like spaces had an appearance suggestive of Kaposi’s sarcoma. The tumor focally appeared to recapitulate splenic sinuses in nine cases, creating a pseudosinusoidal pattern blended with the slit-like vascular channels. One case had features suggestive of lymphatic origin. The lesion was characterized by eosinophilic proteinaceous fluid filled vascular spaces associated with foamy macrophages, papillary structures lined by malignant cells that projected into the lumina, and a sparse lymphoid infiltrate in the supporting connective tissue (Fig. 4).

A, slit-like spaces simulate splenic cords and sinuses. B, anastomosing vascular channels give a ‘honeycomb-like’ appearance.

A, capillary vascular channels. B, fascicles of spindled cells reminiscent of fibrosarcoma.

Angiosarcoma with features suggestive of lymphatic origin (lymphangiosarcoma) with eosinophilic proteinaceous fluid and papillary structures lined by malignant cells projecting into vascular lumina.
A solid sarcomatous pattern often interfaced with the vasoformative regions (n = 19). The sarcomatous constituent resembled fibrosarcoma in two cases (Fig. 3B) and a malignant fibrous histiocytoma in one case. Papillary projections with hyaline cores covered by malignant endothelial cells could be seen protruding into the vascular lumina or cystic spaces in many tumors (n = 14) (Fig. 5). Sheets of malignant epithelioid cells with abundant opaque to eosinophilic cytoplasm and round to irregular nuclei with vesicular nuclear chromatin, and variably accentuated nucleoli, were conspicuous in five cases (Fig. 6). Hemophagocytosis by the malignant cells (n = 8) and intracytoplasmic hyaline globule clusters (n = 12) could be readily identified (Fig. 7A). Interestingly, six of the eight cases with hemophagocytosis also had hyaline globules (highlighted with a periodic acid-Schiff). Mitotic figures were identified in most cases (n = 23), ranging from less than 1 to 9/10 high-power fields (HPF), with and average of 4/10 HPF. Furthermore, hemorrhage (n = 28), necrosis (n = 23), hemosiderin (n = 18), fibrosis (n = 15), extramedullary hematopoiesis (n = 8, Fig. 7B), amyloid (n = 1, Fig. 5), and calcification (n = 1) was found to a variable degree. The patient with amyloid deposition had no history of a lymphoproliferative disease. The two long-term survivors (cases 9 and 19) failed to demonstrate any distinguishing histologic features. Case 9 was characterized by alternating slit-like and honeycomb-like spaces lined by endothelial cells showing mild atypia, and one mitotic figure/10 HPF. In contrast, case 19 was comprised of endothelial cells showing moderate atypia forming slit-like to cavernous spaces alternating with solid areas, foci of necrosis, and one mitotic figure/10 HPF.

Papillary structures with hyaline cores lined by malignant cells project into luminal spaces with amyloid present focally.

Angiosarcoma with an epithelioid appearance.

A, hemophagocytosis and phagocytosis of hyaline globules. B, extramedullary hematopoiesis.
Immunohistochemical Profile
Twenty-one of 25 cases (84%) were positive for two or more markers of vascular differentiation (CD31, CD34, and FVIIIRAg) (Table 4) (Fig. 8). Seventeen of the 25 tumors (68%) were immunoreactive for all three markers. The case reminiscent of lymphangiosarcoma (case 4) was positive for CD31 and CD34 and negative for FVIIIRAg and for vascular endothelial growth factor receptor 3 (VEGFR3). Although VEGFR3 was noted in nine of 17 angiosarcomas in our series (53%), all nine of these cases were also immunoreactive for the standard vascular markers: CD31, CD34, and FVIIIRAg. Three of 27 cases were immunoreactive for CD8 (11%). CD21 was negative in all 22 cases studied.

Immunoreactivity of splenic angiosarcoma for VEGFR3.
Markers of histiocytic differentiation (CD68 and lysozyme) were observed in 24 of 27 cases (89%) in which one or both studies were performed. Nine of the 24 cases (38%) were positive for both. The case reminiscent of lymphangiosarcoma was positive for both CD68 and lysozyme. High Ki-67 values (≥10%) were observed in 14 of 25 (56%) cases studied (including the two long-term survivors), but they did not correlate with disease outcome (P = .580).
A similar antibody panel was applied to 10 cases of littoral cell angioma, and nine cases each of splenic hemangioma and lymphangioma (Table 4). All of the tumors were from adult patients. The tumors universally expressed at least two markers of vascular differentiation, and all of the “tall” (histiocyte-like) cells of littoral cell angiomas expressed both markers of histiocytic differentiation. VEGFR3 was immunoreactive in three of six hemangiomas (50%) and all lymphangiomas (100%) and littoral cell angiomas (100%) (Table 4). Surprisingly, both CD68 and lysozyme were detected in six of nine hemangiomas (67%), but in none of the lymphangiomas. The cells of normal splenic sinusoids and endothelial cells were consistently reactive with VEGFR3 as well as FVIIIRAg, CD31, CD8, CD68, and lysozyme. The splenic sinusoidal cells were consistently negative for CD34. In contrast, endothelial cells of small vessels were consistently positive for CD34.
Contributor Diagnoses
The submitting pathologists in 19 of 25 cases (76%) rendered a diagnosis of angiosarcoma. Additional interpretations included agnogenic myeloid metaplasia, peliosis, malignant fibrous histiocytoma, hemangioma, and congestion. A diagnosis was not submitted in three cases.
DISCUSSION
The term angiosarcoma currently encompasses cases accepted as angiosarcomas and lymphangiosarcomas (7, 8). Angiosarcomas of soft tissue are uncommon, and those of the spleen are rare. Although most of our cases occurred in patients older than 40 years and specifically in the seventh decade, patients of any age may be afflicted (2, 3, 9, 10, 11, 12, 13). Women were more likely to present at an older age than men. However, this finding may simply reflect the longevity of women and/or represent a spurious result due to our small population size. Also in our study, there were slightly more men than women, with a ratio of 4:3.
Most patients (21 of 28) suffered abdominal pain. Other presenting symptoms included generalized weakness, fatigue, malaise, and/or fever, similar to the findings documented by other investigators (1, 2, 3, 5, 10, 11, 14, 15, 16, 17). Our observed rate of splenic rupture (25%) is slightly higher than that of other authors (1, 3), perhaps related to the massive splenomegaly observed in our patients. However, splenic rupture was not associated with clinical outcome (P = .840). Anemia was the most common laboratory abnormality, although thrombocytopenia and leukocytosis were also noted, again, findings similar to the literature (1, 2, 3, 5, 9, 10, 11, 15, 17, 18, 19, 20, 21). A variety of explanations can be proffered to explain the anemia, including anemia of chronic disease, bleeding, hemolysis, sequestration of erythroid elements, a tumor associated factor, obliteration of the normal bone marrow hematopoietic elements, and/or old age. The cause is likely multifactorial; however, our results do not suggest a specific answer to this hypothesis.
Similar to earlier observations, all of the patients studied developed metastasis (10, 11, 16, 17, 19, 20, 21). Other investigators report cases of splenic angiosarcoma developing years after radiation therapy for other malignancies, although the spleen was not usually in the radiation field(s) (2, 18). No etiologic association with chemotherapeutic agents has been substantiated in the literature (3), nor in any of our patients, even though one patient had been previously exposed to chemotherapeutic agents. In spite of these various clinical parameters and treatment protocols, our study corroborates findings of other investigators that splenic angiosarcomas are aggressive neoplasms with an extremely grave prognosis. Most patients die within a few years of diagnosis, independent of treatment(s) (1, 2, 3, 4, 10, 11, 12, 14, 18, 19).
The histologic appearance of splenic angiosarcoma is quite heterogeneous. Malignant vascular tumors of the spleen show at least a focal vasoformative component, and in most cases this is the dominant pattern. However, the prominent epithelioid and malignant mesenchymal features observed in some cases emphasize the need to examine multiple sections and perform immunohistochemical analysis. Although mitotic figures are present in most cases, we found architecture and cytologic atypia to be much more helpful in determining the diagnosis because cytologic atypia was identified in all cases studied. Additional features that were helpful to distinguish malignant from benign vascular tumors included foci of necrosis, hemophagocytosis, hyaline globules, and prominent foci of extramedullary hematopoiesis (3).
In spite of all of our cases being immunoreactive for at least one marker of vascular differentiation, the immunoreactivity was occasionally focal and weak, highlighting the utility of applying a panel of antibodies. Among the splenic angiosarcomas, we did not find cytokeratin reactivity as previously described for epithelioid angiosarcomas (22), although one case did react with epithelial membrane antigen (13).
The vascular endothelial growth factor receptor (VEGFR) family is comprised of a group of three tyrosine kinase receptors for a family of vascular endothelial growth factors (23). VEGFR-3 has been found in most endothelia during embryogenesis. Later in development, it becomes restricted to lymphatic endothelium in most tissue (24, 25). It has also been found to be upregulated in the endothelial cells of tumor neovascularization (26) and is consistently expressed in lymphangiomas (25), Kaposi’s sarcoma (24), and in hemangiomas and angiosarcomas (27). Our experience on angiosarcomas show VEGFR3 expression in over one-half of the cases (9 of 17), suggesting a derivation from an immature endothelial-type cell.
A surprising finding was that the majority of our cases (14 of 25) expressed at least one marker associated with histiocytic differentiation (CD68 and/or lysozyme), including all eight cases that showed hemophagocytosis by the malignant cells. Some authors have questioned the utility of CD68 as a marker of histiocytic differentiation, as it is also expressed in some nonhistiomonocytic neoplasms (28). In contrast, lysozyme appears to be restricted to histiocytes and macrophages, submucosal glands, serum, and granulocytes (29, 30). Lysozyme has also been documented specifically in splenic sinusoidal (littoral) endothelial cells and other red pulp lining cells by many authors (31, 32, 33).
Isolated cases in the literature have documented histiocytic differentiation in splenic angiosarcomas (3, 31, 34, 35). This finding may be unique to the spleen, as there are only a couple of reports that demonstrate histiocytic differentiation in angiosarcomas of the soft tissues (35, 36). Reactivity with histiocytic markers suggests the possibility that at least some of these tumors may arise from splenic lining cells, although absorption of lysozyme from serum by the malignant cells cannot be excluded.
Splenic hamartomas and littoral cell angiomas are both considered primary splenic lesions, most likely derived from the splenic red pulp. Similar to the angiosarcomas in our study, both of these tumors show “biphasic” immunoreactivity for vascular and histiocytic markers (31, 32, 33, 37, 38, 39). Further support for the theory that splenic angiosarcomas arise from the splenic red pulp is lent by the finding of only red pulp involvement in three cases. Unlike normal splenic sinusoidal cells and the formative cells of splenic hamartoma, only a minority of the angiosarcomas in the present study was immunoreactive for CD8. However, Buckley et al. (40) have demonstrated phenotypic and anatomically distinct subsets of cells with histiocytic properties in the normal spleen. Buckley later discovered that cells lining the splenic cords are immunoreactive for CD68 and lysozyme and negative for CD8 (similar to many angiosarcomas in the present study), whereas splenic sinusoidal lining cells (littoral cells) are immunoreactive for lysozyme and CD8 (32). These findings suggest primary splenic angiosarcoma as well as other primary splenic tumors may be derived from subsets of splenic lining cells.
The etiology of splenic angiosarcomas remains elusive, without the site specific predisposing factors and agents of other anatomic locations. These include (but are not limited to) shrapnel, steel, Dacron graft material, surgical sponges, bone wax, thorium dioxide, insecticides, vinyl chloride, and anabolic steroids (3, 41, 42, 43, 44, 45). We were unable to elicit a history of exposure to any of these agents in our cases; however, because most of the patients were dead, the recollections of family members or notes made in the medical record may not have specifically covered all entities.
Case 4 is an unusual vascular malignancy with features that previously have been attributed to lymphangioma, and that have been sometimes observed in cases thought to represent lymphangiosarcoma. These features include eosinophilic protein-rich fluid filled spaces, numerous papillary structures, focal lymphocytic infiltrate between and within the vascular lumens, and immunoreactivity for markers of vascular differentiation (CD31 and FVIIIRAg). Lymphocytes were noted in the papillary projections as well as in the cystic spaces and supporting connective tissue (7, 46, 47, 48, 49). To our knowledge, this is the first reported case of splenic angiosarcoma with these features (MEDLINE 1966 to 1999). At present it is not possible to differentiate between lymphangiosarcoma and angiosarcoma utilizing immunohistochemical studies or electron microscopy, therefore this case is currently best classified as an angiosarcoma (7, 8). Similar to the majority of the other cases, the patient developed multiple metastases and died with tumor 9 mo following presentation.
In summary, splenic angiosarcoma is an aggressive neoplasm that is almost uniformly fatal. The macroscopic appearance varies from one to several nodular, well-circumscribed masses to large ill-defined regions of hemorrhage and necrosis. The microscopic appearance is quite heterogenous, although a vasoformative component can be identified. A panel of immunohistochemical studies to include markers of vascular and histiocytic differentiation may be quite helpful in confirming both the vascular origin of the tumor, as well as histiocytic differentiation, that may be unique to primary splenic angiosarcomas. We propose that most angiosarcomas of the spleen arise from splenic lining cells, although additional studies on a larger cohort of cases are needed to confirm this hypothesis.
References
Wick MR, Smith SL, Scheithauer BW, Beart RW Jr . Primary nonlymphoreticular malignant neoplasms of the spleen. Am J Surg Pathol 1982; 6: 229–242.
Sordillo EM, Sordillo PP, Hajdu SI . Primary hemangiosarcoma of the spleen: report of four cases. Med Pediatr Oncol 1981; 9: 319–324.
Falk S, Krishnan J, Meis JM . Primary angiosarcoma of the spleen. A clinicopathologic study of 40 cases. Am J Surg Pathol 1993; 17: 959–970.
McGinley K, Googe P, Hanna W, Bell J . Primary angiosarcoma of the spleen: a case report and review of the literature. South Med J 1995; 88: 873–875.
Smith VC, Eisenberg BL, McDonald EC . Primary splenic angiosarcoma. Case report and literature review. Cancer 1985; 55: 1625–1627.
Hsu SM, Raine L, Fangor H . Use of avidin-biotin-peroxidase techniques in comparison between ABC and unlabeled antibody (PAP) procedures. J Histochem Cytochem 1981; 29: 577–580.
Enzinger FM, Weiss SW . Malignant vascular tumors. In: Enzinger FM, Weiss SW, editors. Soft tissue tumors. 3rd ed. New York: Mosby; 1994. p. 641–655.
Kindblom LG, Stenman G, Angervall L . Morphological and cytogenetic studies of angiosarcoma in Stewart-Treves syndrome. Virchows Arch A Pathol Anat Histopathol 1991; 419: 439–445.
Chen K, Bolles CG, Gilbert EF . Angiosarcoma of the spleen: a report of two cases and review of the literature. Arch Pathol Lab Med 1979; 103: 122–124.
Garvin DF, King FM . Cysts and nonlymphomatous tumors of the spleen. Pathol Annu 1981; 16: 61–80.
Alt B, Hafez GR, Trigg M, Shahidi NT, Gilbert EF . Angiosarcoma of the liver and spleen in an infant. Pediatr Pathol 1985; 4: 331–339.
Naka N, Ohsawa M, Tomita Y, Kanno H, Uchida A, Myoui A, et al. Prognostic factors in angiosarcoma: a multivariate analysis of 55 cases. J Surg Oncol 1996; 61: 170–176.
Meis-Kindblom JM, Kindblom LG . Angiosarcoma of soft tissue: a study of 80 cases. Am J Surg Pathol 1998; 22: 683–697.
DeNavasquez S . Angioblastoma of the spleen with metastasis in the liver. J Pathol 1936; 42: 651–656.
Nagasue N, Ogawa Y, Inokuchi K . Hemangiosarcoma of liver and spleen treated by hepatic artery ligation, intraportal infusion chemotherapy, and splenectomy. Cancer 1976; 38: 1386–1390.
Yucel AE, Durak H, Bernay I, Bayraktar Y, Bekdik C, Telatar H . Functional asplenia and portal hypertension in a patient with primary splenic hemangiosarcoma. Clin Nucl Med 1990; 15: 324–326.
Hayasaka K, Saitoh Y, Imanoto T, Takashio T, Mineta M, Kikuchi Y, et al. Case report of malignant splenic tumor. Radiat Med 1992; 10: 65–69.
Wilkinson HA3, Lucas JC, Foote FW Jr . Primary splenic angiosarcoma. A case report. Arch Pathol 1968; 85: 213–218.
VanDerburgh AJ . Primary hemangiosarcoma of the spleen with sarcomatosis. J Am Osteopath Assoc 1975; 74: 510–514.
Kaneko K, Onitsuka H, Murakami J, Honda H, Kimura M, Shiraishi N, et al. MRI of primary spleen angiosarcoma with iron accumulation. J Comput Assist Tomogr 1992; 16: 298–300.
Ha HK, Kim HH, Kim BK, Han JK, Choi BI . Primary angiosarcoma of the spleen. Acta Radiol 1994; 35: 455–458.
Wenig BM, Abbondanzo SL, Heffess CS . Epithelioid angiosarcoma of the adrenal glands. A clinicopathologic study of nine cases with a discussion of the implications of finding “epithelial-specific” markers. Am J Surg Pathol 1994; 18: 62–73.
Korpelainen EI, Alitalo K . Signaling angiogenesis and lymphangiogenesis. Curr Opin Cell Biol 1998; 10: 159–164.
Jussila L, Valtola R, Partanen TA, Salven P, Heikkila P, Matikainen MT, et al. Lymphatic endothelium and Kaposi’s sarcoma spindle cells detected by antibodies against the vascular endothelial growth factor receptor-3. Cancer Res 1998; 58: 1599–1604.
Lymboussaki A, Partanen TA, Olofsson B, Thomas-Crusells J, Fletcher CD, de Waal RM, et al. Expression of the vascular endothelial growth factor C receptor VEGFR-3 in lymphatic endothelium of the skin and in vascular tumors. Am J Pathol 1998; 153: 395–403.
Valtola R, Salven P, Heikkila P, Taipale J, Joensuu H, Rehn M, et al. VEGFR-3 and its ligand VEGF-C are associated with angiogenesis in breast cancer. Am J Pathol 1999; 154: 1381–1390.
Partanen TA . Lack of lymphatic vascular specificity of vascular endothelial growth factor receptor 3 in 185 vascular tumors. Cancer 1999; 86: 2406–2412.
McHugh M, Miettinen M . KP1 (CD68), it’s limited specificity for histiocytic tumors. Appl Immunohistochem 1994; 186–190.
Mason DY, Taylor CR . The distribution of muramidase (lysozyme) in human tissues. J Clin Pathol 1975; 28: 124–132.
Franken C, Meijer CJ, Dijkman JH . Tissue distribution of antileukoprotease and lysozyme in humans. J Histochem Cytochem 1989; 37: 493–498.
Arber DA, Strickler JG, Chen YY, Weiss LM . Splenic vascular tumors: a histologic, immunophenotypic, and virologic study. Am J Surg Pathol 1997; 21: 827–835.
Buckley PJ . Phenotypic subpopulations of macrophages and dendritic cells in human spleen. Scanning Microsc 1991; 5: 147–157.
Buckley PJ, Dickson SA, Walker WS . Human splenic sinusoidal lining cells express antigens associated with monocytes, macrophages, endothelial cells, and T lymphocytes. J Immunol 1985; 134: 2310–2315.
Meybehm M, Fischer HP . Littoralzellangiosarkom der Milz. Morphologische, immunhistochemische Befunde und Uberlegungen zur Histogenese eines seltenen Milztumors. Pathologe 1997; 18: 401–405.
Takato H, Iwamoto H, Ikezu M, Kato N, Ikarashi T, Kaneko H . Splenic hemangiosarcoma with sinus endothelial differentiation. Acta Pathol Jpn 1993; 43: 702–708.
Hitchcock MG, Hurt MA, Santa Cruz DJ . Cutaneous granular cell angiosarcoma. J Cutan Pathol 1994; 21: 256–262.
Falk S, Stutte HJ, Frizzera G . Littoral cell angioma, a novel splenic vascular lesion demonstrating histiocytic differentiation. Am J Surg Pathol 1991; 15: 1023–1033.
Falk S, Stutte HJ . Hamartomas of the spleen: a study of 20 biopsy cases. Histopathology 1989; 14: 603–612.
Zukerberg LR, Kaynor BL, Silverman ML, Harris NL . Splenic hamartoma and capillary hemangioma are distinct entities: immunohistochemical analysis of CD8 expression by endothelial cells. Hum Pathol 1991; 22: 1258–1261.
Buckley PJ, Smith MR, Braverman MF, Dickson SA . Human spleen contains phenotypic subsets of macrophages and dendritic cells that occupy discrete microanatomic locations. Am J Pathol 1987; 128: 505–520.
Creech JLJ, Johnson MN . Angiosarcoma of liver in the manufacture of polyvinyl chloride. J Occup Med 1974; 16: 150–151.
Horta JS . Late effects of thorotrast on the liver and spleen, and their efferent lymph nodes. Ann N Y Acad Sci 1967; 145: 676–699.
Fawcett FJ, Easterbrook P, Smerdon GR . Angiosarcoma of liver and spleen in a scrap metal merchant. Br J Ind Med 1983; 40: 113–114.
Mori T, Sakai T, Nuzue Y, Okamoto T, Wada T, Tanaka T, et al. Malignancy and other injuries following Thorotrast administration: follow-up study of 147 cases in Japan. Strahlentherapie 1967; 134: 229–254.
Popper H, Thomas LB, Telles NC, Falk H, Selikoff IJ . Development of hepatic angiosarcoma in man induced by vinyl chloride, thorotrast, and arsenic. Comparison with cases of unknown etiology. Am J Pathol 1978; 92: 349–369.
Sordillo PP, Chapman R, Hajdu SI, Magill GB, Golbey RB . Lymphangiosarcoma. Cancer 1981; 48: 1674–1679.
Tomita K, Yokogawa A, Oda Y, Terahata S . Lymphangiosarcoma in postmastectomy lymphedema (Stewart-Treves syndrome): ultrastructural and immunohistologic characteristics. J Surg Oncol 1988; 38: 275–282.
Gebhart M, Chasse E, Petein M . Lymphangiosarcoma. Reports of 3 cases and review of the literature. Eur J Surg Oncol 1995; 21: 211–214.
Cerri A, Gianni C, Corbellino M, Pizzuto M, Moneghini L, Crosti C . Lymphangiosarcoma of the pubic region: a rare complication arising in congenital non-hereditary lymphedema. Eur J Dermatol 1998; 8: 511–514.
Acknowledgements
The authors wish to thank R. Allen Frommelt, Ph.D., for his skilled statistical analysis and Robin-Anne Ferris, M.F.S., for her expert photography.
Author information
Authors and Affiliations
Corresponding author
Additional information
The opinions or assertions contained herein are the private views of the authors and are not to be construed as official or as reflecting the views of the Department of the Air Force, the Department of the Navy, or the Department of Defense.
Rights and permissions
About this article
Cite this article
Neuhauser, T., Derringer, G., Thompson, L. et al. Splenic Angiosarcoma: A Clinicopathologic and Immunophenotypic Study of 28 Cases. Mod Pathol 13, 978–987 (2000). https://doi.org/10.1038/modpathol.3880178
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.3880178
Keywords
This article is cited by
-
Imaging features of primary sites and metastatic patterns of angiosarcoma
Insights into Imaging (2021)
-
Primary Splenic Angiosarcoma with Liver Metastasis Caused by Malign Transformation of Hemangioma: A Case Report and Literature Review
Journal of Gastrointestinal Cancer (2021)
-
Benign Lesions of the Spleen—Are They Really So Benign? A Report of Two Cases
Indian Journal of Surgery (2020)
-
A case of angiosarcomas which occurred in an adrenal gland and spleen synchronously
International Cancer Conference Journal (2018)
-
Well-differentiated angiosarcoma of spleen: a teaching case mimicking hemagioma and cytogenetic analysis with array comparative genomic hybridization
World Journal of Surgical Oncology (2015)
