
Abstract
Lymph node metastasis is one of the strongest negative prognostic factors for patients with Barrett’s adenocarcinoma (BCA). However, despite the importance of the metastatic process in BCA, the molecular basis of it remains poorly understood. To search for cytogenetic events associated with metastasis in regional or distant lymph nodes in BCA, we investigated 8 primary BCA and their lymph node metastases and compared them with 18 nonmetastatic BCA. In metastatic primary BCA, we observed significantly more DNA gains on 3q (P = .013), 17q (P = .019), and 22q (P = .021) compared with nonmetastatic primary BCA. No statistically significant correlation could be observed between DNA copy number changes and the histopathologic stage, grade, or survival (P > .05). The most frequent alteration observed only in lymph node metastases but not in the related primary tumor was loss of 2q (5 of 8). Coamplification of 7p and chromosome 17 was found in 6 of 8 lymph node metastases. A comparison of DNA copy number changes between primary tumors and their corresponding metastases indicated a high degree of genetic heterogeneity. Fluorescence in situ hybridization analysis demonstrated the involvement of the Her-2/neu gene in primary BCA and its related lymph node metastases. Each of the investigated primary tumors and related lymph node metastases also showed striking heterogeneity with respect to Her-2/neu, with several areas displaying different levels of amplification. In summary, our data indicate that DNA copy number changes on 2q, 3q, 7p, 17q, and 22q may be involved in the metastatic process in BCA. Furthermore, the striking genetic heterogeneity that we found between primary BCA and its lymph node metastases may underlie BCA’s poor responsiveness to therapy and could help explain why prognostic biomarkers measured exclusively in primary tumors give an incomplete view of the biologic potential of BCA.
Similar content being viewed by others


INTRODUCTION
Barrett’s adenocarcinoma (BCA) has risen faster in incidence than any other gastrointestinal tumor over the past 30 years, and it has a poor prognosis (1). In particular, the presence of lymph node metastasis has been shown to be an independent indicator of poor prognosis in several studies of BCA (2, 3). Unfortunately, approximately 30 to 60% of patients present with lymph node metastases at the time of initial diagnosis and have an overall 5-year survival rate of less than 10%. The identification of patients who are at greatest risk for harboring or developing metastases could help in the design of new strategies for the diagnosis and management of this disease. In addition, the identification of genes responsible for metastasis may allow the development of new specifically targeted therapeutic regimens.
Many studies have focused on the genetic changes that occur along the proposed metaplasia-dysplasia-carcinoma sequence of Barrett’s esophagus, but little is known regarding the genetic steps that promote the later stages of tumor progression and the development of metastatic phenotype. Cytogenetic studies using G-banding, fluorescence in situ hybridization (FISH), and comparative genomic hybridization (CGH) have revealed a complex pattern of structural and numeric chromosomal aberrations in adenocarcinoma of the distal esophagus and gastric cardia (4, 5, 6, 7, 8). In recent years, a complete allelotype of BCA has been constructed (9, 10, 11). Loss of heterozygosity in BCA is seen most frequently at 4q, 5q, 9p, 12q, 13q, 17p, and 18q. Recently, CGH studies of adenocarcinomas of the gastroesophageal junction have revealed gains on chromosomes 20, 6p, 7p, 7q, 8q, and 17q and losses on 4p, 4q, 5q, and 18q in BCA (6, 7, 8, 12). However, the relevance of these and other genetic alterations for the emergence of the metastatic phenotype is unknown, despite the crucial importance of this event for the prognosis of patients who have BCA.
To search for the cytogenetic events related to the metastatic process, we investigated and compared metastatic BCA and its corresponding nodal metastases with nonmetastatic BCA using CGH to evaluate chromosome copy number changes, followed by FISH analysis for validation and characterization of the amplifications on 17q that were observed at a significantly higher frequency in metastatic tumors and their metastases. In addition, the analysis of paired samples from the same patient made it possible to asses the degree of clonal divergence and genetic heterogeneity, which is an important basis for the understanding of the biology of metastasis in BCA as a means for the development of improved strategies for prognostic assessment and treatment.
MATERIALS AND METHODS
Patient Samples
Twenty-six patients (one female, 25 males) who had BCA of the distal esophagus that was diagnosed between 1990 and 1998 were studied. Follow-up data were available for 24 cases (mean follow-up, 22 months; range, 0 to 85 months). All patients underwent an esophagectomy without preoperative radiotherapy or chemotherapy. The presence of distant metastasis was excluded preoperatively in these patients by means of a chest x-ray, computer tomography of the thorax and abdomen, percutaneous ultrasound, and bone scintillography. Postoperative follow-up also included these procedures at regular intervals to exclude the possibility of tumor recurrence and/or distant metastases. The primary tumors were staged according to the Union Internationale Contre le Cancer TNM system (13). Nineteen BCA were classified as pT1, 2 as pT2, and 5 as pT3. Clinical and histopathologic data for the study group are summarized in Table 1. Formalin-fixed and paraffin-embedded tissue blocks of 18 nonmetastatic (17 pT1 and 1 pT3) and 8 metastatic (2 pT1, 2 pT2, and 4 pT3) BCA and 8 corresponding nodal metastases (1 celiac axis lymph node and 7 regional lymph nodes) were selected for laser microdissection and subsequent DNA extraction.
Laser-Assisted Microdissection and Cell Pretreatment
An ultraviolet laser microbeam (P.A.L.M, Wolfratshausen, Germany) was used to excise tumor cell groups from defined tissue areas on unmounted hematoxylin and eosin–stained serial sections (5 μm). From each case of BCA, 105 to 106 microdissected tumor cells were sampled, representing the complete carcinomatous area of one to three serial sections. At least 1 to 5 × 103 microdissected cells from three to six serial sections were sampled from regional lymph node metastases and normal squamous epithelium (control). The cells were lysed in 50 to 200 μl 100 mm Tris-HCl (pH 7.5), 1 mg/mL proteinase K for 24 h at 55° C.
Amplification and Labeling of Test DNA
Degenerate oligonucleotide primed polymerase chain reaction (DOP-PCR) was performed on DNA extracts of the microdissected tissue according to a previously published method (14, 15, 16). DOP-PCR amplified DNA as well as nonamplified DNA from tumor and control samples was labeled with biotin-16-dUTP (Boehringer Mannheim, Mannheim, Germany) using a standard nick translation reaction.
CGH and Image Analysis
CGH was performed on test DNA amplified by DOP-PCR according to published procedures (14, 15, 16). For all CGH preparations, 300 ng labeled test DNA and SpectrumRed direct-labeled normal female or male total human genomic DNA (Vysis, Inc., Downers Grove, U.K.), plus 25 μg CotIDNA were cohybridized to denatured metaphases for 72 h at 37° C. After hybridization, biotin-labeled test DNA was detected with Cy2-conjugated streptavidin (Dianova, Hamburg, Germany). For CGH analysis, at least 10 metaphases were imaged and karyotyped after visualization with a Zeiss Axioplan 2 fluorescence microscope (Zeiss, Oberkochen, Germany) equipped with filter sets (single-band excitation filters) for 4′-6-diamidino-2-phenylindole, Cy2, and Texas Red. Averaged profiles were generated by CGH analysis software (ISIS 3, V2.84; MetaSystems, Altlussheim, Germany) from at least 10 to 15 homologous chromosomes. Based on previous CGH experiments (14, 17, 18) the ratios 1.25 and 0.75 were used as diagnostic cutoff levels indicating overrepresentation (DNA amplification) and underrepresentation (DNA loss), respectively.
Control Experiments
DOP-PCR amplified DNA obtained from morphologically normal appearing esophageal squamous epithelium (n = 6) was hybridized with nonamplified reference DNA (SpectrumRed) to metaphase preparations. In these experiments, no chromosomal changes were detected except for chromosomal regions 1p34–36 and 19. These regions are known to show artifactual results by CGH (15, 19). Therefore, chromosomes 1p and 19 were excluded from further analysis. In addition, three cases of BCA were comprehensively analyzed using both DOP-PCR amplified and nonamplified DNA, with the same chromosomal changes being detected by both methods. CGH results were further validated by comparison with FISH analysis.
FISH Analysis
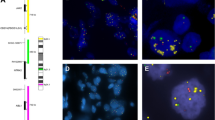
FISH analysis with specific probes was essential to validate and further characterize the CGH findings. Moreover, FISH analysis allowed visualization of the degree of intratumoral heterogeneity. Five metastatic BCA and six related metastases, as well as two nonmetastatic BCA specimens with DNA copy number changes on chromosomes 17q, known from the CGH experiments, were selected to validate these changes (Table 2). Serial 5-μm sections of the tissue blocks were used for FISH analysis, which investigated areas corresponding to those examined by CGH. For FISH analysis, a PathVysion HER-2 DNA probe kit (Vysis) was used according to the manufacturer’s recommendations. The kit consists of directly labeled fluorescent DNA probes specific for the HER-2/neu gene locus (17q11.2-q12) and a DNA probe specific for the α satellite DNA sequence at the centromeric region of chromosome 17 (17p11.1-q11.1). Signals from 100 to 150 tumor cell nuclei per specimen were counted using confocal laser scanning microscopy (Zeiss LSM 510). Nuclei from normal squamous epithelium or lymphocytes deposited separately on the same slide were used as controls of hybridization efficiency and specificity. The criteria established by Hopman et al. (20) were followed for signal enumeration. The Her-2/neu gene locus was classified as amplified if there were more than twice as much red Her-2/neu signals than green centromere 17 signals (ratio more than 2). More than two red signals accompanied by the same number of green signals was considered to be indicative of polysomy of chromosome 17. Her-2/neu clusters were defined as an accumulation of usually more than 10 signals. In those cells, the precise number of signals could not be counted because of coalescent signal clusters (Fig. 1A). When the proportion of cells with negative nuclei exceeded 20%, the procedure was regarded as suboptimal and was either repeated or abandoned.

A, bubble plots for paired comparison of amplification frequencies of 37 chromosomal arms in metastatic and nonmetastatic BCA. The bubble sizes represent multiplicity of pairs with identical frequency values for different chromosome arms, in ascending order: 1, 2, 3, 4, and 7 pairs. The dotted lines represent diagonal and 95% limits based on Fisher’s exact test. 1) 4p, 5p, 10p, 14q, 18q; 2) 5q, 12q; 3) 9p, 21q, Y. B, bubble plots for paired comparison of deletion frequencies of 37 chromosomal arms in metastatic and nonmetastatic BCA. The bubble sizes represent multiplicity of pairs with identical frequency values for different chromosome arms, in ascending order: 1, 2, 3, 4, and 7 pairs. The dotted lines represent diagonal and 95% limits based on Fisher’s exact test. 1) 3q, 6p, 7p, 8q, 11p, 11q, 20p, 20q; 2) 1q, 2p, 18p, 22q; 3) 12p, 17q; 4) 3p, 21q; 5) 8p, 12q.
Statistical Analysis
The statistical significance of differences between the number of DNA gains and losses of primary and metastatic tumors and the histopathologic (pTNM, grade) and clinical (survival) data was calculated using Wilcoxon’s rank-sum test and Fisher’s exact test, respectively. For comparison of average aberration frequencies in each entity, the standard error of the mean (±SEM) was calculated. Correlation with recurrent cytogenetic findings was analyzed using Fisher’s exact test.
RESULTS
Overview of Copy Number Changes
DNA gains and losses determined by CGH for the 8 metastatic BCA, including their associated regional lymph node metastases, and the 18 nonmetastatic BCA are shown in Tables 3 and 4. All of the specimens showed DNA copy number changes by CGH. The total number of chromosomal aberrations was 13.1 ± 1.4 in the 26 primary tumors. A comparison of the chromosomal aberrations with histopathologic (pTNM, grade) and clinical (survival) data revealed no statistically significant correlation (P > .05).
CGH of Metastatic Primary BCA
An average of 16.0 ± 3.6 chromosomal imbalances per case were detected in the eight metastatic BCA. The chromosomal alterations most often identified were gains on 8q and 20q (75% each); 2p and 3q (63% each); 7p and 17q (50% each); 6p and 7q (38% each); and 11q, 16q, and 22q (each 37%). Losses were observed on the Y chromosome and on 5q (75% each); 4q (63%); 4p, 9p, 13q, and 14q (50% each); and 6q, 7q, and 18q (37% each). Within the metastatic tumor group, there was no statistically significant correlation between chromosomal aberrations and pT category (pT1 versus pT2; pT2 versus pT3; pT1 versus pT3; all P > .05).
CGH of Lymph Node Metastasis
In the eight lymph node metastases, an average of 21.1 ± 1.3 chromosomal imbalances per case were detected. These consisted of gains on 8q and 20q (100%); 7p and 17q (88% each); 2p, 3p, 6p, and 16p (75% each); and 9q, 10p, 10q, and 16q (62% each) and losses on 4q (87%); 2q, 5q, and 13q (75% each); Y chromosome (50%); 9p and 15q (50% each); and 18q, 14q, and 6q (38% each). Coamplification of chromosome 7p and 17 was found in 6 of 8 metastases but in only 3 of 8 of the primary tumors of these cases. The most frequent alteration observed only in metastases but not in the related primary BCA was loss of 2q (with a minimal common region at 2q22–23), which was detected in 5 of 8 metastases (Table 5).
CGH of Nonmetastatic Primary BCA
An average of 11.8 ± 1.1 chromosomal imbalances per case were detected in the 18 nonmetastatic BCA cases. The chromosomal alterations most often identified were gains on 8q (83%), 20q (61%), 7p (56%), 10q (55% each), 2p and 6p (44% each), and 13q (39%). Losses were observed predominantly on the Y chromosome (78%), 9p (50%), 18q (44%), and 5q and 7q (33% each).
CGH Comparison of Metastatic and Nonmetastatic Primary BCA
There was a slightly but not significantly higher total number of chromosomal imbalances in metastatic BCA (16.0 ± 3.6) compared with nonmetastatic BCA (11.8 ± 1.1). In particular, we observed significantly more DNA gains on 3q (P = .013), 17q (P = .019), and 22q (P = .021) in the primary tumors of the metastatic group. There was no detectable deletion that was significantly associated with the metastatic phenotype. Figure 1 presents a paired comparison of the amplification (A) and deletion (B) frequencies of 37 chromosome arms in the metastatic and nonmetastatic BCA cases. No substantial differences were observed in the number of chromosomal imbalances between metastatic and nonmetastatic BCA concerning the pT staging.
CGH Comparison of Paired Samples of Primary Tumors and Their Metastases
The total number of chromosomal imbalances in the lymph node metastases (21.1 ± 1.3) was higher than that of the related primary tumors (16.0 ± 3.6) but was not statistically significantly different (P > .05). Of the aberrations that appeared as new changes in the metastases, the most frequent was loss of 2q (5 of 8 metastases); however, there were no aberrations that were significantly more frequent in nodal metastases than in primary carcinomas. The degree of similarity between changes in the primary tumor and its metastasis varied widely. None of the pairs had identical genetic changes. A comparison of the DNA copy number changes in the primary tumors and their related metastases is presented in Table 5.
FISH Analysis in Paired Samples of Primary Tumors and Their Metastases
A total of 13 FISH experiments on five metastatic primary BCA, six related regional lymph node metastases, and two nonmetastatic BCA were performed. A comparison of the results from FISH and CGH analysis is presented in Table 2. The fraction of cells with amplified Her-2/neu in each tumor was calculated, the results of which are shown in Table 2. Three primary BCA and three lymph node metastases displayed distinct clusters of Her-2/neu signals. In these cases, the precise number of signals could not be counted, but the clusters were usually composed of more than 10 signals. Several scored tumor cells contained equal numbers of centromere 17 and Her-2/neu signals. Each of the investigated tumors showed striking heterogeneity, with several areas displaying different levels of Her-2/neu. Intratumoral heterogeneity was further indicated by the presence of polysomic cells that were not amplified in these tumors. Figure 2 illustrates intratumoral heterogeneity of Her-2/neu amplification in a primary BCA (Fig. 2A) and in its lymph node metastasis (Fig. 2B, C).

Heterogeneity of Her-2/neu gene amplification in case E16 in a primary BCA (A) and its lymph node metastasis (B and C) shown by dual-color FISH with Her-2/neu-specific probes (red signals) and chromosome 17 centromeric probes (green signals) counterstained with 46′-diamidino-2-phenylindole-2 Hcl. Note the tumor cells displaying different levels of HER-2/neu amplification indicating intratumoral genetic heterogeneity. The tumor cells on the right side demonstrate coalescent Her-2/neu signal clusters (red signals). Several scored tumor cells contained equal numbers of centromere 17 and Her-2/neu signals, indicative of polysomy of chromosome 17. In the regional lymph node metastasis, most cells showed clusters, as shown in C; however, the signal distribution as shown in B was also found.
DISCUSSION
The present study demonstrates that certain genetic alterations are significantly more frequent in metastatic than in nonmetastatic BCA. Furthermore, identified were chromosomal aberrations that preferentially occur in lymph node metastases, suggesting that they may be involved in the metastatic process.
Primary BCA are genetically complex tumors, as shown by the large number of genetic changes per case in our series (mean, 13.1 ± 1.4). Many of the common genetic changes detected in our series have been reported in previous cytogenetic studies (4, 5, 6, 7, 8). Most of the alterations found in the nonmetastatic primary tumors were also commonly found in the metastatic BCA cases. In particular, the statistical analysis indicated that DNA gain on chromosomes 3q, 17q, and 22q was significantly more frequent in metastatic BCA (P < .05). No significant correlation could be observed between pT stage and chromosomal aberrations, indicating that DNA gains on 3q, 17q, and 22q are specifically involved in the metastatic process. Thus, these chromosomal changes may be potential targets for candidate genes important for metastasis.
Overrepresentation of chromosome arm 3q was observed in 5 of 8 primary tumors of the metastatic BCA, with a minimal common region of 3q26-qter, but in only 2 of 18 nonmetastatic BCA. Increased copy number changes at 3q26–27 have also been reported as a recurrent change in a variety of tumors that are widely known for early development of their metastatic potential, such as small cell carcinoma of the lung (21). One potentially relevant gene at 3q26.3 whose product may contribute to the control of cell proliferation and malignant transformation is PIK3CA. This gene encodes the catalytic subunit of phosphatidylinositol-3 kinase, a critical component of several cell signaling pathways, including those of epidermal growth factor, platelet-derived growth factor, and insulin-like growth factor (22).
DNA gain on chromosome 17q was also observed in a significantly higher proportion of metastatic than nonmetastatic primary BCA. An association between chromosome 17 gains and lymph node metastasis has been demonstrated in both breast and gastric cancers (23, 24). Some cell regulation and growth factor genes have been assigned to the area of 17q12–21 amplification. Of these, the best potential candidate genes are Her-2/neu and Grb7, the former being known to be overexpressed in gastric cancer (25, 26). The Grb7 is a newly identified SH2-containing protein that avidly binds to activated EGFR (27). It localizes to 17q11–12 near the Her-2/neu locus. Stein et al. demonstrated frequent coamplification of Grb7 and Her-2/neu in breast carcinoma cells (28). Furthermore, Tanaka et al. showed that Grb7 isoforms are involved in cell invasion and metastatic progression of esophageal squamous carcinoma (29). Thus, Grb7 may also be involved in tumor progression of BCA.
DNA gain on chromosome 22q was significantly more frequent in metastatic primary BCA than in nonmetastatic primary BCA. Previous CGH studies of adenocarcinomas of the stomach and the gastroesophageal junction have revealed gain on 22q in a low frequency (7, 30, 31). In addition, colorectal and gastric carcinomas frequently have loss of heterozygosity on chromosome 22q, suggesting that inactivation of tumor suppressor genes on 22q participates in the tumor development (32). However, to our knowledge, no potential oncogene located on 22q is known to be involved in tumorigenesis of carcinomas of the digestive tract.
However, CGH is limited in its ability to detect DNA losses and amplifications, because the use of metaphase chromosomes limits detection of events involving small genomic regions (of less than 20 Mb) and resolution of closely spaced aberrations. In particular, CGH is less sensitive than other techniques in detecting genetic loss; thus, it is possible that some of the genetic deletion events that were not statistically correlated with metastasis in our study may prove to be using other methodologies, especially for loci with genetic loss that nearly reached significant values (4p-, 5q-, 6q-). Therefore, these results should be confirmed with other techniques (e.g., high-resolution CGH microarrays, loss of heterozygosity analysis) in further studies.
By combining CGH and FISH results, we were able to demonstrate that HER-2/neu gene amplification was present in both primary BCA and the lymph node metastases in our series. Hardwick et al. (33) showed, by immunohistochemical staining, that HER-2/neu overexpression is not involved in the early stages of neoplastic progression in Barrett’s esophagus but plays a role in late events such as metastatic progression. Nevertheless, our study group was too small to clarify the clinical relevance of HER-2/neu in BCA progression, although we did demonstrate a statistically significantly higher proportion of 17q gain in metastatic versus nonmetastatic BCA.
An intriguing finding in the regional lymph node metastases was the coamplification of 7p and chromosome 17 in 6 of 8 cases, whereas this coamplification was present in only 3 of 8 of the primary tumors. In a previous study of BCA, coamplification of the EGFR gene (located on 7p12) and Her-2/neu was observed in 15.4% of cases (34). Coamplification of Her-2/neu and EGFR has also been reported in gastric adenocarcinoma (35), and it has been suggested that a synergistic selection for multiple copies of both the Her-2/neu and EGFR genes may occur during progression to cancer (36). Whether coamplification of these genes occurs in more aggressive esophageal adenocarcinoma awaits further studies. The most frequent alteration that was observed in our series of metastases but was not seen in the related primary BCA was the loss of 2q (with a minimal common region at 2q22–23) detected in 5 of 8 metastases. A potential candidate gene in this area is Grb14, which is assigned to the chromosomal region 2q22–24. The Grb14 gene is a member of Grb7 gene family discussed above (37).
The combination of the findings of the CGH and FISH analyses in paired primary tumors and nodal metastases allowed us to study the clonal relationship between the primary tumors and their metastases. None of the pairs had identical chromosomal changes, but all shared at least some alterations. Although most sample pairs showed a high degree of commonality, some metastases had little evidence of clonal relationship to their primary tumor. One possible explanation for this finding is the selection pressure of the tissue growth environment. Growth conditions may differ between the primary and metastatic sites (38), providing a growth advantage to different cell clones in the two areas. The low degree of shared chromosomal change seen by CGH also suggests that a clonal relationship might be missed as a result of genetic heterogeneity within the primary tumor. The development of divergent clones during the process of clonal expansion is proposed to be the explanation for tumor cell heterogeneity (38). Such an intratumoral heterogeneity was demonstrated by the FISH analysis of the primary BCA tumors. Each of the investigated carcinomas was composed of several areas with different centromeric 17/Her2-neu signals, indicating a high prevalence of intratumoral heterogeneity (Table 2 and Fig. 2). Even if limited concordance between primary BCA and their metastases were seen, this does not necessarily conflict with the model of histologic progression. Barrett et al. (39) demonstrated using Barrett’s esophagus cell lines that clonal evolution is more complex than predicted by linear models.
In conclusion, using a combined CGH and FISH analysis technique, we have described genetic aberrations associated with the metastatic behavior of BCA. Different genetic alterations were seen in primary BCA with and without metastases, which points to potential chromosomal regions that could harbor genes responsible for tumor progression and metastasis. In particular, our data suggest that chromosomes 2q, 3q, 7p, and 17q all are worthy of further analysis with more focused techniques. Moreover, we demonstrate that considerable genetic heterogeneity exists within the primary tumor and within related metastases in BCA, as well as between primary BCA and its metastases. This finding helps to explain the poor responsiveness of BCA to therapy and is a factor that indicates that biomarkers of prognosis measured exclusively in primary tumors may be limited in their ability to describe the biologic potential of BCA.
Note: Since the writing of this article, we have had the opportunity to investigate the metaplasia-dysplasia-carcinoma sequence in our BCA cases (40). Interestingly, distinct patterns of chromosomal aberrations were already present in the preinvasive stages of BCA.
References
Cameron AJ, Lomboy CT, Pera M, Carpenter HA . Adenocarcinoma of the esophagogastric junction and Barrett’s esophagus. Gastroenterology 1995; 109: 1541–1546.
Streitz JM Jr, Ellis FH Jr, Gibb SP, Balogh K, Watkins E Jr . Adenocarcinoma in Barrett’s esophagus. A clinicopathologic study of 65 cases. Annu Surg 1991; 213: 122–1225.
Paraf F, Flejou JF, Pignon JP, Fekete F, Potet F . Surgical pathology of adenocarcinoma arising in Barrett’s esophagus. Analysis of 67 cases. Am J Surg Pathol 1995; 19: 183–191.
Menke-Pluymers MB, van Drunen E, Vissers KJ, Mulder AH, Tilanus HW, Hagemeijer A . Cytogenetic analysis of Barrett’s mucosa and adenocarcinoma of the distal esophagus and cardia. Cancer Genet Cytogenet 1996; 90: 109–117.
Persons DL, Croughan WS, Borelli KA, Cherian R . Interphase cytogenetics esophageal adenocarcinoma and precursor lesions. Cancer Genet Cytogenet 1998; 106: 11–17.
Moskaluk CA, Hu J, Perlman EJ . Comparative genomic hybridization of esophageal and gastroesophageal adenocarcinomas shows consensus areas of DNA gain and loss. Genes Chromosomes Cancer 1998; 22: 305–311.
El-Rifai W, Harper JC, Cummings OW, Hyytinen ER, Frierson HF Jr, Knuutila S, et al. Consistent genetic alterations in xenografts of proximal stomach and gastro-esophageal junction adenocarcinomas. Cancer Res 1998; 58: 34–37.
van Dekken H, Geelen E, Dinjens WN, Wijnhoven BP, Tilanus HW, Tanke HJ, et al. Comparative genomic hybridization of cancer of the gastroesophageal junction: deletion of 14Q31–32.1 discriminates between esophageal (Barrett’s) and gastric cardia adenocarcinomas. Cancer Res 1999; 59: 748–752.
Gleeson CM, Sloan JM, McGuigan JA, Ritchie AJ, Weber JL, Russell SEH . Barrett’s oesophagus: microsatellite analysis provides evidence to support the proposed metaplasia-dysplasia-carcinoma sequence. Genes Chromosomes Cancer 1998; 21: 49–60.
Barrett MT, Galipeau PC, Sanchez CA, Emond MJ, Reid BJ . Determination of the frequency of loss of heterozygosity in esophageal adenocarcinoma by cell sorting, whole genome amplification and microsatellite polymorphisms. Oncogene 1996; 12: 1873–1878.
Hammoud ZT, Kaleem Z, Cooper JD, Sundaresan RS, Patterson GA, Goodfellow PJ . Allelotype analysis of esophageal adenocarcinomas: evidence for the involvement of sequences on the long arm of chromosome 4. Cancer Res 1996; 56: 4499–4502.
van Dekken H, Vissers CJ, Tilanus HW, Tanke HJ, Rosenberg C . Clonal analysis of a case of multifocal esophageal (Barrett’s) adenocarcinoma by comparative genomic hybridization. J Pathol 1999; 188: 263–266.
Hermanek P, Wittekind LH . Classification of malignant tumors. Berlin: Springer-Verlag; 1997.
Zitzelsberger H, Kulka U, Lehmann L, Walch A, Smida J, Aubele M, et al. Genetic heterogeneity in a prostatic carcinoma and associated prostatic intraepithelial neoplasia as demonstrated by combined use of laser-microdissection, degenerate oligonucleotide primed PCR and comparative genomic hybridization. Virchows Arch 1998; 433: 297–304.
Weber RG, Scheer M, Born IA, Joos S, Cobbers JM, Hofele C, et al. Recurrent chromosomal imbalances detected in biopsy material from oral premalignant and malignant lesions by combined tissue microdissection, universal DNA amplification, and comparative genomic hybridization. Am J Pathol 1998; 153: 295–303.
Lu YJ, Osin P, Lakhani SR, Di Palma S, Gusterson BA, Shipley JM . Comparative genomic hybridization analysis of lobular carcinoma in situ and atypical lobular hyperplasia and potential roles for gains and losses of genetic material in breast neoplasia. Cancer Res 1998; 58: 4721–4727.
Walch AK, Zitzelsberger HF, Aubele MM, Mattis AE, Bauchinger M, Candidus S, et al. Typical and atypical carcinoid tumors of the lung are characterized by 11q deletions as detected by comparative genomic hybridization. Am J Pathol 1998; 153: 1089–1098.
Aubele M, Mattis A, Zitzelsberger H, Walch A, Kremer M, Hutzler P, et al. Intratumoral heterogeneity in breast carcinoma revealed by laser-microdissection and comparative genomic hybridization. Cancer Genet Cytogenet 1999; 110: 94–102.
Kallioniemi OP, Kallioniemi A, Piper J, Isola J, Waldman FM, Gray JW, et al. Optimizing comparative genomic hybridization for analysis of DNA sequence copy number changes in solid tumors. Genes Chromosomes Cancer 1994; 10: 231–243.
Hopman AH, Ramaekers FC, Raap AK, Beck JL, Devilee P, van der Ploeg M, et al. In situ hybridization as a tool to study numerical chromosome aberrations in solid bladder tumors. Histochemistry 1988; 89: 307–316.
Balsara BR, Sonoda G, du Manoir S, Siegfried JM, Gabrielson E, Testa JR . Comparative genomic hybridization analysis detects frequent, often high-level, overrepresentation of DNA sequences at 3q, 5p, 7p, and 8q in human non-small cell lung carcinomas. Cancer Res 1997; 57: 2116–2120.
Volinia S, Hiles I, Ormondroyd E, Nizetic D, Antonacci R, Rocchi M, et al. Molecular cloning, cDNA sequence, and chromosomal localization of the human phosphatidylinositol 3-kinase p110-alpha (PIK3CA) gene. Genomics 1994; 24: 472–477.
Herrington CS, Leek RD, McGee JO . Correlation of numerical chromosome 11 and 17 imbalance with metastasis of primary breast cancer to lymph nodes. J Pathol 1995; 176: 353–359.
Terada R, Yasutake T, Yamaguchi E, Hisamatsu T, Nakamura S, Ayabe H, et al. Higher frequencies of numerical aberrations of chromosome 17 in primary gastric cancers are associated with lymph node metastasis. J Gastroenterol 1999; 34: 11–17.
Park JB, Rhim JS, Park SC, Kimm SW, Kraus MH . Amplification, overexpression, and rearrangement of the erbB-2 protooncogene in primary human stomach carcinomas. Cancer Res 1989; 49: 6605–6609.
Tahara E . Molecular biology of gastric cancer. World J Surg 1995; 19: 484–488; discussion 489–490.
Margolis B, Silvennoinen O, Comoglio F, Roonprapunt C, Skolnik E, Ullrich A, et al. High-efficiency expression/cloning of epidermal growth factor-receptor-binding proteins with Src homology 2 domains. Proc Natl Acad Sci U S A 1992; 89: 8894–8898.
Stein D, Wu J, Fuqua SA, Roonprapunt C, Yajnik V, D’Eustachio P, et al. The SH2 domain protein GRB-7 is co-amplified, overexpressed and in a tight complex with HER2 in breast cancer. EMBO J 1994; 13: 1331–1340.
Tanaka S, Mori M, Akiyoshi T, Tanaka Y, Mafune K, Wands JR, et al. A novel variant of human Grb7 is associated with invasive esophageal carcinoma. J Clin Invest 1998; 102: 821–827.
Nessling M, Solinas-Toldo S, Wilgenbus KK, Borchard F, Lichter P . Mapping of chromosomal imbalances in gastric adenocarcinoma revealed amplified protooncogenes MYCN, MET, WNT2, and ERBB2. Genes Chromosomes Cancer 1998; 23: 307–316.
Kokkola A, Monni O, Puolakkainen P, Larramendy ML, Victorzon M, Nordling S, et al. 17q12–21 amplicon, a novel recurrent genetic change in intestinal type of gastric carcinoma: a comparative genomic hybridization study. Genes Chromosomes Cancer 1997; 20: 38–43.
Muraoka M, Konishi M, Kikuchi-Yanoshita R, Tanaka K, Shitara N, Chong JM, et al. p300 gene alterations in colorectal and gastric carcinomas. Oncogene 1996; 12: 1565–1569.
Hardwick RH, Shepherd NA, Moorghen M, Newcomb PV, Alderson D . c-erbB-2 overexpression in the dysplasia/carcinoma sequence of Barrett’s oesophagus. J Clin Pathol 1995; 48: 129–132.
al-Kasspooles M, Moore JH, Orringer MB, Beer DG . Amplification and over-expression of the EGFR and erbB-2 genes in human esophageal adenocarcinomas. Int J Cancer 1993; 54: 213–219.
Yoshida K, Tsuda T, Matsumura T, Tsujino T, Hattori T, Ito H, et al. Amplification of epidermal growth factor receptor (EGFR) gene and oncogenes in human gastric carcinomas. Virchows Arch B Cell Pathol Incl Mol Pathol 1989; 57: 285–290.
Yin XY, Donovan-Peluso M, Whiteside TL, Johnson JT, Day R, Herberman RB, et al. Gene amplification and gene dosage in cell lines derived from squamous cell carcinoma of the head and neck. Genes Chromosomes Cancer 1991; 3: 443–454.
Daly RJ, Sanderson GM, Janes PW, Sutherland RL . Cloning and characterization of GRB14, a novel member of the GRB7 gene family. J Biol Chem 1996; 271: 12502–12510.
Nowell PC . The clonal evolution of tumor cell populations. Science 1976; 194: 23–28.
Barrett MT, Sanchez CA, Prevo LJ, Wong DJ, Galipeau PC, Paulson TG, et al. Evolution of neoplastic cell lineages in Barrett oesophagus. Nat Genet 1999; 22: 106–109.
Walch A, Zitzelsberger H, Bruch J, Keller G, Angermeier D, Aubele M, et al. Chromosomal imbalances in Barrett’s adenocarcinoma and in the metaplasia-dysplasia-carcinoma sequence. Am J Pathol 2000; 156: 555–566.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Walch, A., Zitzelsberger, H., Bink, K. et al. Molecular Genetic Changes in Metastatic Primary Barrett’s Adenocarcinoma and Related Lymph Node Metastases: Comparison with Nonmetastatic Barrett’s Adenocarcinoma. Mod Pathol 13, 814–824 (2000). https://doi.org/10.1038/modpathol.3880143
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.3880143
Keywords
This article is cited by
-
Discordance in HER2 Status in Gastro-esophageal Adenocarcinomas: A Systematic Review and Meta-analysis
Scientific Reports (2017)
-
Functional Polymorphisms Associated with Disease-Free Survival in Resected Carcinoma of the Esophagus
Journal of Gastrointestinal Surgery (2011)
-
Genome-wide analysis of genetic alterations in Barrett's adenocarcinoma using single nucleotide polymorphism arrays
Laboratory Investigation (2009)
-
Œsophage de Barrett, dysplasie, adénocarcinome: histomorphologie et apport de la biologie moléculaire
Acta Endoscopica (2008)
-
Frequent homogeneous HER-2 amplification in primary and metastatic adenocarcinoma of the esophagus
Modern Pathology (2007)
