
Abstract
Melanoma antigen recognized by T cells 1 (MART-1) and tyrosinase-related protein-2 (TRP-2) are two useful markers for immunohistochemical detection of melanocytic tumors. However, these markers may be passively acquired (phagocytosed) rather than actively synthesized. Reverse transcriptase in situ polymerase chain reaction (RT in situ PCR) can amplify even small amounts of specific mRNA in cells and therefore confirm the cellular source of a marker. We developed a one-step RT in situ PCR procedure in which Thermus thermophilus DNA polymerase synthesizes and amplifies cDNA from mRNA in a single reaction mixture. To examine its practicability and feasibility with formalin-fixed, paraffin-embedded (FFPE) tissue, we compared the results of one-step RT in situ PCR with those of immunohistochemistry (IHC). MART-1 mRNA was identified in the cytoplasm of lesional cells from 23/26 primary melanomas (92%), 9/9 metastatic melanomas (100%) and 5/6 nevi (83%). MART-1 epitope was detected by IHC in 23/24 primary melanomas (96%), 9/9 metastatic melanomas (100%) and 5/6 nevi (83%). TRP-2 mRNA was identified in the cytoplasm of lesional cells from 17/26 primary melanomas (65%), 6/9 metastatic melanomas (67%) and 4/6 nevi (67%). TRP-2 epitope was detected by IHC in 20/24 primary melanomas (83%), 9/9 metastatic melanomas (100%) and 4/6 nevi (67%). Both techniques detected MART-1 and TRP-2 in FFPE melanoma cell lines. Neither marker was detected in squamous cell carcinomas or basal cell carcinomas by RT in situ PCR or IHC. We conclude that the RT in situ PCR technique can be successfully applied to FFPE tissue to determine the cellular sources of gene expression observed by conventional PCR approaches.
Similar content being viewed by others

Main
The ability to detect gene expression at the mRNA level in single identifiable cells would enhance our understanding of the molecular basis of biological responses. The reverse transcriptase in situ polymerase chain reaction (RT in situ PCR) may be used to detect mRNA in cells, in tissue sections, cytospins or imprints. Unlike in situ hybridization, which also localizes specific nucleic acid sequences at the cellular level, RT in situ PCR has an amplification step that makes it more sensitive for low endogenous levels of mRNA.
In previous studies of the RT in situ PCR technique, we demonstrated the presence of mRNA for the melanoma markers tyrosinase (TYR) and MAGE-3 (MAGEA3) in melanoma cell lines but not in nonmelanoma cell lines.1, 2 We successfully modified the RT in situ PCR approach to detect mRNA for tyrosinase in archived formalin-fixed, paraffin-embedded (FFPE) tissues.3 However, although the technique used in these early studies has been applied widely, its technical complexity and unreliability prevent routine diagnostic application in FFPE archival materials.4
We have developed a one-step RT-PCR procedure that uses a single Thermus thermophilus DNA polymerase (Tth DNA polymerase) to detect and amplify mRNA for melanoma markers in melanoma cell lines and archival FFPE tissues of melanocytic and other skin tumors. In the one-step RT-PCR, the Tth DNA polymerase synthesizes gene-specific cDNA from the target mRNA sequence in the presence of Mn2+ at elevated temperatures and amplifies the cDNA in the same reaction. We used this procedure to detect melanoma antigen recognized by T cells 1 (MART-1, also known as MELAN-A (MLANA))5, 6, 7 and tyrosinase-related protein-2 (TRP-2, also known as DOPAchrome tautomerase (DCT)).8, 9 These two melanoma-associated genes are expressed with high specificity in normal melanocytes and melanocytic lesions, including melanoma and nevi. We used the same specimens to compare the sensitivity and accuracy of our one-step RT in situ PCR technique with the sensitivity and accuracy of immunohistochemistry (IHC) with antibodies to MART-1 and TRP-2.
Materials and methods
Cell Block Preparation
Three melanoma cell lines (M7, M14 and M26) were obtained from the John Wayne Cancer Institute at Saint John's Hospital and Health Center (Santa Monica, CA, USA) (courtesy Dr DL Morton). The cell lines were established and characterized at UCLA.10, 11, 12 The cultured cells were grown in RPMI-1640 plus 10% penicillin and streptomycin (Gibco, Grand Island, NY, USA), harvested and collected in a Falcon tube. The cells were pelleted by centrifugation at 1500 r.p.m. for 10 min, washed three times with phosphate-buffered saline (PBS) and resuspended immediately in neutral-buffered formalin for 6–8 h. After fixation, the cells were centrifuged at 1500 r.p.m. for 10 min, washed in sterile distilled water and alcohols graded from 75 to 100%. The cell pellet was suspended for 2 h in xylene and embedded in paraffin wax at 65°C in a tissue cassette applied to a microtome support.
Archival Tissues from Melanomas, Nevi and other Tumors
FFPE tissue specimens were retrieved from the archives of the Surgical Pathology Section at UCLA and the Klinikum Nürnberg Nord, Nürnberg, Germany, with Institutional Review Board permission from both institutions. Of the 54 blocks, 35 were from melanoma (20 invasive primary melanomas, 6 melanomas in situ and 9 melanoma metastases), 6 were from melanocytic nevi, 8 were from nonmelanocytic cutaneous tumors (4 squamous cell carcinomas and 4 basal cell carcinomas) and 5 were from tumor-free axillary lymph nodes from patients with breast cancer. Conventionally stained slides from all tissues were reviewed to confirm the diagnosis.
Immunohistochemistry
Representative sections from each lesion were processed for immunohistochemical staining with a monoclonal antibody cocktail for MART-1 (M2-7C10/M2-9E3; Zymed Laboratories, South San Francisco, CA, USA) and a goat polyclonal antibody for TRP-2 (Santa Cruz Biotechnology, Santa Cruz, CA, USA).
Tissue sections (4 μm thick) were deparaffinized in xylene and dehydrated in graded ethanols. Epitope retrieval was performed by boiling in citrate buffer (pH 6.0) for 30 min. Endogenous peroxidase activity was blocked by 3% hydrogen peroxide for 30 min. Primary antibodies were diluted in PBS at 1:20 for MART-1 and 1:100 for TRP-2. Sections were incubated with the primary antibodies overnight at 4°C. Antibody-bridge labeling used the streptavidin–biotin–peroxidase method with biotinylated anti-mouse antibody (Vector Laboratories, Burlingame, CA, USA) or biotinylated anti-goat antibody (DakoCytomation, Glostrup, Denmark) and streptavidin horseradish peroxidase (Zymed Laboratories). Sections were then reacted with aminoethyl carbazole as a chromogen (AEC; Zymed Laboratories), counterstained with hematoxylin and mounted.
RT In Situ PCR
Preparation and pretreatment
Tissue sections (4 μm thick) were placed on silane-coated slides, deparaffinized and hydrated through xylene and a graded alcohol series. The slide-mounted tissue sections were digested with trypsin (0.2 or 0.4 mg/ml; Roche Diagnostics, Mannheim, Germany) at 37°C for 30–60 min and treated with RNase-free DNase I (70 U per tissue section; Roche Diagnostics) at 37°C overnight.
One-step RT in situ PCR
The one-step RT in situ PCR approach was modified from a previously described technique.13, 14 The oligonucleotide primer sequences (Invitrogen, Carlsbad, CA, USA) were derived from a previous paper15 as shown in Table 1. For RT-PCR of MART-1 mRNA, the forward primer, 5′-CACGGCCACTCTTACACCAC-3′, and the reverse primer, 5′-GGAGCATTGGGAACCACAGG-3′, yielded a product of 254 bp. For RT-PCR of TRP-2 mRNA, the forward primer, 5′-GAGGTGCGAGCCGACACAAG-3′, and the reverse primer, 5′-CGGTGCCAGGTAACAAATGC-3′, yielded a product of 476 bp. The GeneAmp EZ rTth (recombinant Tth) RNA PCR Kit (Applied Biosystems, Foster City, CA, USA) was employed. Each slide was loaded with the following mixture: 10 μl of 5 × EZ rTth buffer, 5 μl of Mn(OAc)2, 1.6 μl each of dATP/dCTP/dGTP/dTTP, 2 μl of Tth DNA polymerase (Applied Biosystems), 0.6 μl digoxigenin-11-dUTP, 1.6 μl of 2% bovine serum albumin (BSA), 0.9 μl of RNase inhibitor (Roche Diagnostics), 1.5 μl of each primer and 20.5 μl diethylpyrocarbonate (DEPC)-treated water. The slides were covered with HybriWell sealing covers (Research Products International, Mount Prospect, IL, USA) and placed on the GeneAmp In Situ PCR System 1000 thermal cycler (PerkinElmer, Foster City, CA, USA). The cDNA syntheses were performed at 60°C for 30 min. After denaturation at 94°C for 3 min, the cDNAs were amplified by 30 cycles of annealing at 55°C for 1 min and denaturation at 94°C for 30 s. The slides were washed in 0.2 × saline-sodium citrate/BSA (BSA; Sigma, St Louis, MO, USA) at 60°C for 15 min and rinsed in Tris-buffered saline three times for 5 min.
Immunodetection of RT-PCR products
The digoxigenin-labeled PCR products were detected after incubation with an alkaline phosphatase–anti-digoxigenin conjugate (1:200) for 30 min (Roche Diagnostics) and developed in a chromogen of nitroblue tetrazolium and 5-bromo-4-chloro-3-indophosphatase (NBT/BCIP) for 15 min (Zymed Laboratories).
Controls
Control preparations were prepared by (1) omission of the primer and (2) omission of the Tth DNA polymerase.
Results
Melanoma Cell Lines
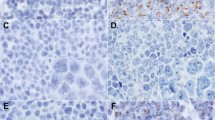
After RT in situ PCR, cytoplasmic mRNA for MART-1 (Figure 1a) and TRP-2 (Figure 1b) was strongly expressed and readily visible on microscopy of the sections prepared from all three melanoma lines (M7, M14 and M26). Nuclear staining was only occasionally observed. Control preparations without the primers (Figure 1c) or without the Tth DNA polymerase (Figure 1d) gave negative results, with no evidence of mRNA amplification.

Specific expression of MART-1 mRNA and TRP-2 mRNA in M26 melanoma cell line. A positive signal is indicated by dark blue to purple coloration, predominantly in the cell cytoplasm. Cytoplasmic mRNA for MART-1 (a) and TRP-2 (b) is strongly expressed. Nuclear DNA was removed by DNase pretreatment. All negative controls, including omission of the primers (c) and omission of the Tth DNA polymerase (d), gave negative results.
Formalin-Fixed, Paraffin-Embedded Tissues
Results from evaluation of 49 archival tissues are summarized in Table 2. The specific and amplified reaction signals detected by RT in situ PCR were mainly located in the cytoplasm.
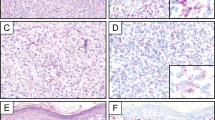
MART-1 epitope was detected by IHC in 5/6 nevi (83%) (Figure 2a), in 32/33 melanomas (97%) (Figure 3a) and in normal basal-layer melanocytes of the follicular and interfollicular epidermis in all skin specimens. RT in situ PCR detected MART-1 mRNA in 23/26 primary melanomas (92%): 6/6 melanomas in situ (radial growth phase melanoma) (100%) and 18/20 invasive melanomas (radial and/or vertical growth phase) (90%) (Figure 3b). MART-1 mRNA was also identified by RT in situ PCR in 9/9 (100%) metastatic melanomas and 5/6 (83%) nevi. Nests of nevus cells diffusely expressed MART-1 mRNA, as did single melanocytes in the epidermis overlying the dermal component of nevi (Figure 2b). The melanocytic nevus that was negative for MART-1 epitope and mRNA was a dermal nevus consisting exclusively of type C nevus cells. Squamous and basal cell carcinomas showed no evidence of MART-1 epitope by IHC or MART-1 mRNA by RT in situ PCR.

Detection of MART-1 in FFPE archival melanocytic nevus tissues. (a) IHC reveals that MART-1 (red coloration) is strongly expressed in nevus cells and single melanocyte in the basal layer of the epidermis. (b) By the RT in situ PCR technique, the reaction product of MART-1 mRNA (dark blue to purple coloration) is predominantly located in the cytoplasm of nevus cells, and the nucleus contains less reaction product. Single melanocytes in the basal layer of the epidermis express MART-1 mRNA (arrows).

Detection of MART-1 and TRP-2 in FFPE archival primary melanoma tissues. (a) IHC reveals that MART-1 (red coloration) is heterogeneously expressed in primary melanoma cells. (b) By the RT in situ PCR technique, the reaction product of MART-1 mRNA (dark blue to purple coloration) is predominantly located in the cytoplasm of melanoma cells. Staining intensity is varied among individual tumor cells. (c) IHC reveals that TRP-2 (red coloration) is strongly expressed in the cytoplasm of melanoma cells. (d) TRP-2 mRNA (dark blue to purple coloration) is also strongly expressed in the cytoplasm of melanoma cells.
TRP-2 epitope was detected by IHC in 20/24 primary melanomas (83%) (5/6 melanomas in situ (83%) and 15/18 invasive melanomas (83%)), 9/9 metastatic melanomas (100%) and 4/6 melanocytic nevi (67%). TRP-2 mRNA was detected by RT in situ PCR in 17/26 primary melanomas (65%) (4/6 melanomas in situ (67%) and 13/20 invasive melanomas (65%)), 6/9 metastatic melanomas (67%) and 4/6 melanocytic nevi (67%). Melanoma cells diffusely expressed TRP-2 (Figure 3c) and TRP-2 mRNA (Figure 3d). Squamous and basal cell carcinomas expressed neither TRP-2 epitope by IHC nor TRP-2 mRNA by RT in situ PCR.
In an invasive melanoma that was negative for MART-1 and TRP-2 epitopes by IHC, RT in situ PCR detected mRNA for MART-1 and TRP-2 in melanoma cells at the dermo-epidermal junction and in the superficial papillary dermis but not in melanoma cells in the deeper dermis.
Slides from five axillary lymph nodes from breast cancer patients showed no evidence of amplification of MART-1 mRNA or TRP-2 mRNA.
Discussion
RT in situ PCR of FFPE tissue sections identified MART-1 mRNA and TRP-2 mRNA in lesional cells of primary and metastatic melanomas and melanocytic nevi. Neither marker was found in cutaneous squamous and basal cell carcinomas, tumors of nonmelanocytic histogenesis.
MART-1, a melanoma antigen recognized by tumor-infiltrating T lymphocytes in melanoma patients,5, 6 is widely used in the evaluation of melanomas. The antibody to MART-1 is specific for the gp100/pmel 17 glycoprotein antigenic group. MART-1 is expressed in normal cells of melanocytic lineage in skin, uveal tract and retina, but is absent from normal epithelia, fibroblasts and lymphocytes. MART-1 is detectable in a majority of melanomas and nevi,16 but is absent from nonmelanocytic neoplasms, with the possible exception of adrenal cortical tumors and gonadal steroidal tumors.17, 18
TRP-2, a melanosomal enzyme with DOPAchrome tautomerase activity, is involved in melanogenesis.8 TRP-1 and TRP-2 are associated with distinct patterns of melanocyte distribution and function in normal skin and cutaneous pigmented lesions.19, 20 A recent study using quantitative PCR and cDNA array hybridization of tissues prepared by laser pressure catapulting verified high expression of TRP-2 by nevi and melanomas.21 TRP-2 is involved in the protection of melanoma cells from apoptosis.22 TRP-1 and TRP-2 also suppress tyrosinase-mediated cell death of melanocytes and melanoma cells.23 Even in later-stage amelanotic melanomas with severely impaired melanin synthesis, pigment-related genes including TRP-1 and TRP-2 are expressed to a variable degree.15
In our study, RT in situ PCR and IHC gave comparable results for detection of MART-1, but respective rates for detection of TRP-2 in primary melanomas were 67 and 83%. The lower sensitivity of RT in situ PCR most likely reflects technical difficulties associated with PCR amplification in situ.4 On the other hand, the intensity of MART-1 and TRP-2 expression by IHC may vary between individual cells in different areas of an individual tumor. Metastatic melanoma, in particular, often shows heterogeneous expression of MART-1.24, 25, 26 The heterogeneous nature of expression of these melanoma-associated antigens expression is related to antigen silencing during melanoma progression. Oncostatin M27 secreted by melanoma cells28 downregulates MART-1 and TRP-2. Microphthalmia transcription factor-M (MITF-M), a melanocyte-specific master transcription factor, may also regulate MART-1 expression.29, 30 Reduced expression of MITF-M is associated with lack of MART-1 expression by melanoma cells. In our study, a melanoma that was negative for MART-1 and TRP-2 by IHC had evidence of MART-1 mRNA and TRP-2 mRNA in melanoma cells at the dermo-epidermal junction and in the superficial papillary dermis, but not in the deep dermis. Thus, antigen silencing can occur during local tumor progression and may permit tumor cells to escape immune recognition and destruction by cytotoxic T cells.31 The clear discrepancy between mRNA and protein levels within some tumor cells is a strong argument for a gene-based method of detection.
The sensitivity and reproducibility of RT in situ PCR can be increased by attention to several technical details. First, the duration of formalin fixation can affect RNA integrity.32, 33, 34 Quantitative RT-PCR analysis has demonstrated MART-1 mRNA in paraffin-embedded melanoma samples up to 3 weeks after fixation. However, detection of total mRNA is markedly reduced in fixed tissues as compared with fresh tissues,35 and prolonged fixation is associated with suboptimal detection of mRNA by RT in situ PCR.
Second, the optimal duration of protease digestion should be determined for each tissue sample (30–60 min in this study).13 Available proteinases include proteinase K, pepsin, pronase and trypsin.36, 37 To avoid overdigestion, we used trypsin, which is more manageable than stronger proteases such as proteinase K. The mRNA signal detected by RT in situ PCR is mainly in the cell cytoplasm. A false-positive signal in the cell nucleus can be prevented by DNase digestion for up to 16 h; longer digestion can damage cellular structure and reduce signal amplification.3, 38 Tissue must be adequately digested by protease before incubation with DNase; otherwise genomic DNA and histone protein will crosslink during formalin fixation, blocking access to DNA by DNase. Nonspecific amplification of residual DNA will create a ‘repair’ artifact.13
A third consideration is the design of the primer. Because we used a specific oligonucleotide primer pair for both reverse transcription and PCR amplification, optimal annealing temperatures for reverse transcription and cDNA amplification were within a close range. Primer pairs can be prepared from the spliced sequences of mRNA to avoid nonspecific PCR products. The use of primers across intron–exon junctions may circumvent problems of amplification of genomic DNA. The MART-1 primers were designed to amplify across four different exons so that the amplified fragment would be only 254 bp, as compared with 16 kb between the same primer sites in the genomic DNA. The TRP-2 primers amplified across three different exons to create an amplicon of 476 bp, as compared with 12 kb between the primer pair in the genomic DNA. During PCR, the 1-min annealing step is long enough for cDNA priming but not for chain-reaction amplification of long genomic DNA sequences. This should prevent the generation of byproducts from genomic DNA. However, the size of amplified product may affect the sensitivity of detection. In our study, the less favorable sensitivity of RT in situ PCR for detection of TRP-2 might have reflected the slightly larger size of the amplicon. Taking into consideration that its sensitivity and reproducibility may still fall short of IHC, further work is required to standardize the procedure for routine use.
There are many potential practical applications of RT in situ PCR. Sentinel lymph node tissue that is negative by histology and immunohistology may have RT-PCR evidence of possible occult metastases.39, 40, 41, 42 However, because of the inevitable cell destruction during RNA extraction, solution-phase RT-PCR cannot identify the cellular source of any amplified signals for mRNA. In this study, we have shown that it is possible to detect MART-1 mRNA and TRP-2 mRNA in archival FFPE tissue sections of melanoma by a one-step RT in situ PCR technique. The in situ detection of melanoma-associated genes may have considerable potential to identify occult metastatic melanoma cells in nodal tissue sections and exclude other sources of melanoma-associated mRNA, such as melanophages.
References
Li PX, Cheng L, Wen DR, et al. Demonstration of cytoplasmic tyrosinase mRNA in tissue-cultured cells by reverse transcription (RT) in situ polymerase chain reaction (PCR) and RT PCR in situ hybridization. Diagn Mol Pathol 1997;6:26–33.
Guo J, Wen DR, Huang RR, et al. Detection of multiple melanoma-associated markers in melanoma cell lines by RT in situ PCR. Exp Mol Pathol 2003;74:140–147.
Guo J, Cheng L, Wen DR, et al. Detection of tyrosinase mRNA in formalin-fixed, paraffin-embedded archival sections of melanoma, using the reverse transcriptase in situ polymerase chain reaction. Diagn Mol Pathol 1998;7:10–15.
Qian X, Lloyd RV . Recent developments in signal amplification methods for in situ hybridization. Diagn Mol Pathol 2003;12:1–13.
Coulie PG, Brichard V, Van Pel A, et al. A new gene coding for a differentiation antigen recognized by autologous cytolytic T lymphocytes on HLA-A2 melanomas. J Exp Med 1994;180:35–42.
Kawakami Y, Eliyahu S, Delgado CH, et al. Cloning of the gene coding for a shared human melanoma antigen recognized by autologous T cells infiltrating into tumor. Proc Natl Acad Sci USA 1994;91:3515–3519.
Kawakami Y, Eliyahu S, Delgado CH, et al. Identification of a human melanoma antigen recognized by tumor-infiltrating lymphocytes associated with in vivo tumor rejection. Proc Natl Acad Sci USA 1994;91:6458–6462.
Tsukamoto K, Jackson IJ, Urabe K, et al. A second tyrosinase-related protein, TRP-2, is a melanogenic enzyme termed DOPAchrome tautomerase. EMBO J 1992;11:519–526.
Hearing VJ, Tsukamoto K, Urabe K, et al. Functional properties of cloned melanogenic proteins. Pigment Cell Res 1992;5:264–270.
Golub SH, Hanson DC, Sulit HL, et al. Comparison of histocompatibility antigens on cultured human tumor cells and fibroblasts by quantitative antibody absorption and sensitivity to cell-mediated cytotoxicity. J Natl Cancer Inst 1976;56:167–170.
Chee DO, Boddie AW, Roth JA, et al. Production of melanoma-associated antigen(s) by a defined malignant melanoma cell strain grown in chemically defined medium. Cancer Res 1976;36:1503–1509.
Irie RF, Irie K, Morton DL . A membrane antigen common to human cancer and fetal brain tissues. Cancer Res 1976;36:3510–3517.
Nuovo GJ . Co-labeling using in situ PCR: a review. J Histochem Cytochem 2001;49:1329–1339.
Adam PJ, Boyd R, Tyson KL, et al. Comprehensive proteomic analysis of breast cancer cell membranes reveals unique proteins with potential roles in clinical cancer. J Biol Chem 2003;278:6482–6489.
Sarantou T, Chi DD, Garrison DA, et al. Melanoma-associated antigens as messenger RNA detection markers for melanoma. Cancer Res 1997;57:1371–1376.
Busam KJ, Chen YT, Old LJ, et al. Expression of melan-A (MART1) in benign melanocytic nevi and primary cutaneous malignant melanoma. Am J Surg Pathol 1998;22:976–982.
Busam KJ, Iversen K, Coplan KA, et al. Immunoreactivity for A103, an antibody to melan-A (Mart-1), in adrenocortical and other steroid tumors. Am J Surg Pathol 1998;22:57–63.
Fetsch PA, Marincola FM, Abati A . The new melanoma markers: MART-1 and Melan-A (the NIH experience). Am J Surg Pathol 1999;23:607–610.
Pisarra P, Lupetti R, Palumbo A, et al. Human melanocytes and melanomas express novel mRNA isoforms of the tyrosinase-related protein-2/DOPAchrome tautomerase gene: molecular and functional characterization. J Invest Dermatol 2000;115:48–56.
Virador V, Matsunaga N, Matsunaga J, et al. Production of melanocyte-specific antibodies to human melanosomal proteins: expression patterns in normal human skin and in cutaneous pigmented lesions. Pigment Cell Res 2001;14:289–297.
Becker B, Roesch A, Hafner C, et al. Discrimination of melanocytic tumors by cDNA array hybridization of tissues prepared by laser pressure catapulting. J Invest Dermatol 2004;122:361–368.
Nishioka E, Funasaka Y, Kondoh H, et al. Expression of tyrosinase, TRP-1 and TRP-2 in ultraviolet-irradiated human melanomas and melanocytes: TRP-2 protects melanoma cells from ultraviolet B induced apoptosis. Melanoma Res 1999;9:433–443.
Rad HH, Yamashita T, Jin HY, et al. Tyrosinase-related proteins suppress tyrosinase-mediated cell death of melanocytes and melanoma cells. Exp Cell Res 2004;298:317–328.
Kageshita T, Kawakami Y, Hirai S, et al. Differential expression of MART-1 in primary and metastatic melanoma lesions. J Immunother 1997;20:460–465.
Cormier JN, Hijazi YM, Abati A, et al. Heterogeneous expression of melanoma-associated antigens and HLA-A2 in metastatic melanoma in vivo. Int J Cancer 1998;75:517–524.
Jungbluth AA, Busam KJ, Gerald WL, et al. A103: an anti-melan-a monoclonal antibody for the detection of malignant melanoma in paraffin-embedded tissues. Am J Surg Pathol 1998;22:595–602.
Durda PJ, Dunn IS, Rose LB, et al. Induction of “antigen silencing” in melanomas by oncostatin M: down-modulation of melanocyte antigen expression. Mol Cancer Res 2003;1:411–419.
Kurnick JT, Ramirez-Montagut T, Boyle LA, et al. A novel autocrine pathway of tumor escape from immune recognition: melanoma cell lines produce a soluble protein that diminishes expression of the gene encoding the melanocyte lineage melan-A/MART-1 antigen through down-modulation of its promoter. J Immunol 2001;167:1204–1211.
Du J, Miller AJ, Widlund HR, et al. MLANA/MART1 and SILV/PMEL17/GP100 are transcriptionally regulated by MITF in melanocytes and melanoma. Am J Pathol 2003;163:333–343.
Kono M, Dunn IS, Durda PJ, et al. Role of the mitogen-activated protein kinase signaling pathway in the regulation of human melanocytic antigen expression. Mol Cancer Res 2006;4:779–792.
Pawelec G . Tumour escape: antitumour effectors too much of a good thing? Cancer Immunol Immunother 2004;53:262–274.
Stanta G, Schneider C . RNA extracted from paraffin-embedded human tissues is amenable to analysis by PCR amplification. BioTechniques 1991;11:304, 306, 308.
Stanta G, Bonin S . RNA quantitative analysis from fixed and paraffin-embedded tissues: membrane hybridization and capillary electrophoresis. BioTechniques 1998;24:271–276.
Godfrey TE, Kim SH, Chavira M, et al. Quantitative mRNA expression analysis from formalin-fixed, paraffin-embedded tissues using 5′ nuclease quantitative reverse transcription-polymerase chain reaction. J Mol Diagn 2000;2:84–91.
Abrahamsen HN, Steiniche T, Nexo E, et al. Towards quantitative mRNA analysis in paraffin-embedded tissues using real-time reverse transcriptase-polymerase chain reaction: a methodological study on lymph nodes from melanoma patients. J Mol Diagn 2003;5:34–41.
Hully JR . In situ PCR. In: Innis MA, Gelfand DH, Sninsky JJ (eds). PCR Applications: Protocols for Functional Genomics. Academic Press: London, 1999, pp 169–194.
Morel G, Raccurt M . PCR/RT-PCR In Situ: Light and Electron Microscopy. CRC Press: Boca Raton, FL, 2003.
Peters J, Krams M, Wacker HH, et al. Detection of rare RNA sequences by single-enzyme in situ reverse transcription-polymerase chain reaction. High-resolution analyses of interleukin-6 mRNA in paraffin sections of lymph nodes. Am J Pathol 1997;150:469–476.
Bostick PJ, Morton DL, Turner RR, et al. Prognostic significance of occult metastases detected by sentinel lymphadenectomy and reverse transcriptase-polymerase chain reaction in early-stage melanoma patients. J Clin Oncol 1999;17:3238–3244.
Rimoldi D, Lemoine R, Kurt AM, et al. Detection of micrometastases in sentinel lymph nodes from melanoma patients: direct comparison of multimarker molecular and immunopathological methods. Melanoma Res 2003;13:511–520.
Takeuchi H, Morton DL, Kuo C, et al. Prognostic significance of molecular upstaging of paraffin-embedded sentinel lymph nodes in melanoma patients. J Clin Oncol 2004;22:2671–2680.
Abrahamsen HN, Sorensen BS, Nexo E, et al. Pathologic assessment of melanoma sentinel nodes: a role for molecular analysis using quantitative real-time reverse transcription-PCR for MART-1 and tyrosinase messenger RNA. Clin Cancer Res 2005;11:1425–1433.
Acknowledgements
We thank Dr DL Morton of the John Wayne Cancer Institute at Saint John's Health Center, Santa Monica, CA, USA for melanoma cell lines M7, M14 and M26. This work is supported by NIH/NCI Grant P01 CA 29605 (subcontract of John Wayne Cancer Institute).
Author information
Authors and Affiliations
Corresponding author
Additional information
This work was presented in part, in poster form, at the 93rd annual meeting of the United States and Canadian Academy of Pathology, Vancouver, BC, Canada, March 6–12, 2004, and at the 12th annual meeting of the Pan American Society for Pigment Cell Research, Newport Beach, CA, June 24–27, 2004.
Duality of interest
The authors of this article declare no duality of interest.
Rights and permissions
About this article
Cite this article
Itakura, E., Huang, RR., Wen, DR. et al. RT in situ PCR detection of MART-1 and TRP-2 mRNA in formalin-fixed, paraffin-embedded tissues of melanoma and nevi. Mod Pathol 21, 326–333 (2008). https://doi.org/10.1038/modpathol.3801008
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.3801008
