
Abstract
Merkel cell carcinoma is a rare and aggressive form of skin cancer of neuroendocrine origin. Its treatment involves wide excision and radiotherapy but no effective therapy exists for advanced disease. Upregulation of the platelet-derived growth factor receptor family of tyrosine kinases, PDGFRA and KIT, has a crucial role in cancer development. Several studies have shown expression of the tyrosine kinase receptor KIT (CD117) in Merkel cell carcinoma. In this study, we examined the expression and mutational status of KIT and PDGFRA in 14 primary and 18 metastatic Merkel cell carcinoma. The expression of KIT and PDGFRA and their respective ligands, stem cell factor (SCF) and PDGFA, was assessed by immunohistochemistry. In addition, we analyzed KIT exons 9, 11, 13 and 17, and PDGFRA exons 10, 12 and 18 for the presence of activating mutations. We found that only 53% of cases of Merkel cell carcinoma expressed KIT, which was mostly seen as diffuse weak staining, and SCF expression was observed only in 31% of cases. In contrast, 87 and 81% of cases expressed PDGFRA and PDGFA, respectively. We observed coexpression of SCF and KIT in only 5 of 32 cases (16%) whereas 25 of 31 cases (81%) showed coexpression of PDGFRA and its ligand PDGFA. While we documented silent mutations in exon 17 of KIT and exons 10, 12 and 18 of PDGFRA, we were not able to identify any known activating mutations. Our results indicate that there is no correlation between positive immunostaining and occurrence of activating mutations in KIT and PDGFRA. Moreover, the presence of KIT/SCF and PDGFRA/PDGFA coexpression in a proportion of cases may indicate an autocrine/paracrine stimulation loop. We think therefore that imatinib mesylate is less likely to be an effective therapy for Merkel cell carcinoma, unless activating mutations exist in other exons of these receptor kinases.
Similar content being viewed by others

Main
Merkel cell carcinoma is a rare, highly aggressive cutaneous malignancy that is considered the second most common cause of death from non-melanoma skin cancer. The disease is characterized by a high incidence of local recurrence and metastasis, and a high mortality rate.1 Approximately 20–25% of patients have regional lymph node involvement at diagnosis.2 Local recurrence rates have been reported between 4 and 62%,3 with the overall 5-year disease-specific survival rate being slightly over 60%.4 The tumor typically arises in the head, neck or extremities of the elderly as a painless, rapidly growing, firm, violaceous papule or nodule.1 There is a male predominance, with a vast majority of individuals affected being white. Exposure to UV radiation from sunlight and/or immunosuppression have been associated with development of this disease, with the former also implicated in its accompanying frequent genetic alterations.5
In 1972, Merkel cell carcinoma was first described by Cyril Toker6 as ‘trabecular carcinoma’, based on the growth pattern of the tumor cells. In 1978, Tang and Toker7 proposed that the normal neuroendocrine Merkel cell in skin is the probable cell of origin for these lesions, given the numerous membrane-bound dense core neurosecretory granules within the cells of trabecular carcinoma. In 1875, Merkel cells were first described by Friedrich Sigmund Merkel and are thought to be slow-acting mechanoreceptors found in the basal layers of the epidermis.8 However, the histogenesis of this neoplasm has been disputed in the literature. Most authors now believe that Merkel cell carcinoma originate from pluripotent stem cells within the epidermis or adnexa.9, 10, 11, 12, 13, 14
Histologically, Merkel cell carcinomas are dermal and/or subcutaneous tumors with a typical ‘undifferentiated’ appearance. The tumor consists of small round basophilic cells that is arranged in syncytial sheets with high nuclear to cytoplasmic ratios, prominent mitotic activity and scattered apoptosis.14 Extensive areas of necrosis, a homogenous ‘salt and pepper’ chromatin pattern, and crush artifact are common findings.14 Immunohistochemically, the tumor cells demonstrate perinuclear dot-like staining with cytokeratin 20. They also show staining with antibodies for CD56, epithelial membrane antigen (EMA) and markers of neuroendocrine differentiation such as chromogranin A, neurofilament protein, neuron-specific enolase and synaptophysin.15
Recently, expression of CD117/KIT transmembrane tyrosine kinase receptor has been demonstrated in Merkel cell carcinoma.16, 17, 18, 19 KIT is a transmembrane protein belonging to the family of type III receptor tyrosine kinases that also includes PDGFRA.20 Another tumor that commonly expresses KIT is gastrointestinal stromal tumor (GIST).21 Successful treatment of metastatic and unresectable GIST with the ATP-competitive protein kinase inhibitor, imatinib mesylate (Gleevec),22, 23 has prompted a search for activating mutations in KIT and related receptor tyrosine kinases in Merkel cell carcinoma. In vitro studies have shown a decrease in proliferation of Merkel cell carcinoma by addition of Gleevec.24
To investigate the status of these receptor tyrosine kinases in this malignancy, we analyzed 27 cases for expression of KIT and PDGFRA and their ligands, stem cell factor (SCF) and PDGFA, respectively. We also looked for mutations in the commonly mutated exons of KIT in GISTs, namely, exons 9, 11, 13 and 17 and the corresponding exons 10, 12 and 18 of PDGFRA.
Materials and methods
Case Selection
A total of 27 cases reviewed at Stanford University Medical Center comprise the study, and consist of 14 primary and 18 metastatic cases (Table 1). We obtained paired samples of primary and metastatic lesions for five cases. Hematoxylin and eosin-stained sections and immunohistochemical stains, if performed at the time of diagnosis, of each case were reviewed to confirm the original findings and assess for the presence of residual tumor in the blocks. All cases that had local recurrence or metastasis to lymph nodes were considered metastatic disease, for simplicity of classification. All cases were selected prior to staining with KIT/CD117, PDGFRA, SCF and PDGFA. This study was approved by the Stanford University's Institutional Review Board.
Immunohistochemistry
Immunohistochemical studies were performed on conventional 4 μm sections of formalin-fixed paraffin-embedded tissues using a modified immunoperoxidase detection system employing the EnVision System (DAKO Corporation, Carpinteria, CA). Primary antibodies were directed against KIT/CD117, PDGFRA, SCF and PDGFA. Heat-induced antigen retrieval was carried out using a pressure cooker either in citrate (10 mM, pH 6.0) or EDTA (1 mM, pH 8.0) buffer. The KIT/CD117 antibody was obtained from DAKO and used at 1:200 dilution (EDTA). The antibodies against SCF (H-189, dilution 1:100, no antigen retrieval), PDGFRA (C-20, dilution 1:100, citrate) and PDGFA (N-30, dilution 1:25, citrate) were obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA). All primary antibodies were incubated for 30 min at room temperature.
Staining for all four antibodies was primarily cytoplasmic, except for CD117, which tended to be primarily membranous with some cytoplasmic staining. Staining scores were reported as follows: strongly positive=moderate to intense staining of >20% of lesional cells; weakly positive=faint, moderate or intense staining of >5% but <20% of lesional cells; and negative=faint staining of <5% to no staining of lesional cells. Positive and negative internal controls were present in all cases for all antibodies. Basal melanocytes served as internal positive controls for CD117, while sebaceous glands served as internal negative controls. KIT-positive GIST was the external positive control. For SCF staining, external controls were used: SCF-expressing breast carcinoma cells served as positive controls while normal breast epithelial cells on the same slide showed no staining and were negative controls. Eccrine sweat glands, which showed a cytoplasmic/membranous pattern of staining, served as internal positive controls for PDGFRA, while small cutaneous nerves were internal negative controls. Blood vessels showed a membranous/cytoplasmic staining pattern for PDGFA, and served as internal positive controls. The endoneurium of cutaneous nerves served as the internal negative control for PDGFA.
KIT and PDGFRA Mutational Analyses
All cases were evaluated for the presence of activating mutations in KIT and PDGFRA. DNA was extracted from formalin-fixed paraffin-embedded tissue blocks using QIAamp DNA Mini Kit (Qiagen Inc., Valencia, CA, USA) according to the manufacturer's instructions. Depending on the amount of tissue, 5–10 sections (5 μm) were cut from each paraffin block. After isolation, genomic DNA from each case was subjected to PCR for amplification of exons 9, 11, 13 and 17 of KIT and exons 10, 12 and 18 of PDGFRA. DNA extracted from normal tonsil, placenta, lymph node and skin ‘tips’ of excisional biopsies of lesional tissue from patients with Merkel cell carcinoma served as negative controls for mutational analysis, while a KIT-positive GIST sample served as a positive control. The primers were designed to amplify the entire exon including the intron–exon junctions. The primer sequences, corresponding annealing temperatures and expected product sizes are listed in Table 2.25, 26 PCR was performed using 10 mM dNTP mix and Platinum Taq DNA Polymerase (Invitrogen Inc., Carlsbad, CA, USA). The PCR conditions are as follows: the samples were incubated for 4 min at 94°C, followed by 35 cycles of 94°C for 30 s, the relevant annealing temperature for 30 s, 72°C for 30 s, and finally incubated at 72°C for 30 min. The amplified products were identified by agarose gel electrophoresis and purified using QIAquick PCR Purification Kit (Qiagen Inc., Valencia, CA, USA) according to the manufacturer's instructions. Subsequently, these products were sequenced at Stanford Protein and Nucleic Acid Facility. The sequencing reaction was performed using forward primers initially, and in cases where there were mutations the variations were confirmed by sequencing in the reverse direction. Approximately 50% of the samples have been sequenced using both forward and reverse primers. The sequence analysis was carried out using BLAST sequence similarity searches from NCBI and the GeneTool Lite 1.0 software from BioTools Inc. Each ABI sequence was compared to the NCBI human KIT (NM_000222) and PDGFRA (NM_006206) gene nucleotide sequences to assess for the presence of any mutations.
Results
Clinical Findings
Most of the patients were elderly (19 of 27 patients were 70 years of age or older) and male (21 of 27 cases, see Table 1). Our data set comprised of 14 primary tumors and 18 metastatic tumors. Among the primary lesions, six were in the head and neck region while eight were from the upper and lower extremities. Sites of metastatic involvement included skin (usually locally recurrent tumors), regional lymph nodes, salivary glands and bone marrow.
Expression of Receptors KIT and PDGFRA
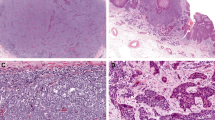
Receptor tyrosine kinase expression was determined in 32 biopsies/excisions of Merkel cell carcinoma from 27 patients (Figure 1). KIT immunoreactivity was observed in 17 of 32 samples (53%), and both membranous and cytoplasmic staining was seen. Four cases (12%) showed strong KIT expression while the remaining 13 cases (41%) expressed KIT weakly (Figure 1c). Unlike KIT, PDGFRA expression was diffuse and cytoplasmic (Figure 1e). Nearly, 87% of cases (27 of 31 cases) showed positive immunostaining for PDGFRA. Of these, 7 cases (23%) showed strong staining and the remaining 20 cases (64%) had weak staining (Table 1).

Immunohistochemical staining with KIT, SCF, PDGFRA and PDGFA in Merkel cell carcinoma. (a) Hematoxylin and eosin (H&E)-stained sections of Merkel cell carcinoma show a nodular and/or diffuse proliferation of uniform basaloid cells within the dermis, extending in some cases to the subcutaneous tissue. (b) High power examination shows cells with ‘salt and pepper’ fine chromatin, high nuclear to cytoplasmic ratios and high mitotic activity. (c–f) KIT staining is primarily membranous (c) while staining for SCF (d) PDGFRA (e) and PDGFA (f) is primarily cytoplasmic.
Expression of Growth Factors SCF and PDGFA
Expression of SCF, the ligand for KIT, was assessed in all cases. Ten cases (31%) expressed SCF in the lesional cells. Of these, only two cases (6%) showed strong cytoplasmic staining (Figure 1d). The remaining eight cases showed diffuse, weak staining. However, significant staining was seen in many cases in the neighboring connective tissues. On the other hand, PDGFA expression was present in 25 of 31 Merkel cell carcinomas (81%), of which 23% (7 cases) showed strong, primarily cytoplasmic staining (Figure 1f). Of the six cases that were negative within the lesional cells, two cases (cases 9 and 30) showed staining in neighboring stromal cells. Interestingly, these cases also showed immunoreactivity for PDGFRA (Table 1).
Polymorphisms Observed in Receptor Tyrosine Kinases
All 32 samples of Merkel cell carcinoma (primary and metastatic lesions) were investigated for mutations in the receptor tyrosine kinases, KIT (exons 9, 11, 13 and 17) and PDGFRA (exons 10, 12 and 18). We largely restricted our analysis to those exons that were frequently mutated in GISTs.27 None of the regions analyzed contained any deletions or insertions. However, we identified what appear to be either mutations or polymorphisms in exon 17 of KIT and in exons 10, 12 and 18 of PDGFRA (Table 3). These mutations/polymorphisms, when present, were the same in both the primary and metastatic lesions for the same patient. For example, primary and metastatic lesions from the same patient (cases 17 and 18) showed similar variations in the PDGFRA exons 10 and 18 sequences. However, there was no correlation between the occurrence of sequence variations and the primary or metastatic status of the tumor. Three cases (11%) revealed a C to T variation at codon 798 in exon 17 of KIT. Both variant codons encode for isoleucine. To check if this was tumor related, we analyzed skin ‘tips’ of an excisional biopsy of a lesion of Merkel cell carcinoma, where the skin ‘tips’ did not contain tumor (case 8). On sequencing, KIT from the skin ‘tips’ did not contain the C to T variation at nucleotide position 2415. Our normal tonsil tissue control also harbored only the wild-type allele.
In addition, two of the Merkel cell carcinoma samples analyzed (7%) showed two mutations in exon 10 of PDGFRA. Of these, one was a previously unreported silent mutation in codon 490 (TTC → TTT). The second variation caused a change in codon 478 from serine to proline which has been reported for malignant peripheral nerve sheath tumors and gliomas.26, 28 These variations were not observed in normal tonsil tissues. All cases analyzed showed a silent mutation in exon 12 of PDGFRA. We observed an A to G change in codon 567 (CCA → CCG), as opposed to the A to T change in the GIST sample we used as the positive control. Analysis of exon 12 of PDGFRA from other normal tissues (tonsil, placenta and lymph node) revealed a similar A to G change at the 1849 nucleotide position. Comparison of this sequence variation with the SNP (single nucleotide polymorphism) database (NCBI) revealed the existence of a known SNP at codon 567 of PDGFRA.
Discussion
Merkel cell carcinoma is a rare and aggressive form of cutaneous malignancy. While the exact route of tumorigenesis is still unknown, several studies have investigated potential pathways of tumor development in this disorder. For example, Van Gele et al29 have shown the presence of missense and nonsense mutations in TP53 in 3 of 15 cases of Merkel cell carcinoma. TP53 and p73 are related genes, which regulate apoptosis, and p73 is thought to be akin to p53 and a candidate tumor suppressor gene. Liu et al30 investigated the expression of β-catenin and the mutational status of genes involved in the Wnt-signaling pathway, and concluded that this pathway did not play a role in the tumorigenesis of Merkel cell carcinoma. The classical MAP kinase signaling pathway was also examined in this skin carcinoma, and found to be not necessarily important, as there were no activating mutations in the B-Raf gene.31
In this study, we evaluated the expression and mutational status of receptor tyrosine kinases, KIT and PDGFRA, in 32 cases of Merkel cell carcinoma. Expression of KIT was observed in 17 of 32 cases (53%). This is a smaller number of positive cases than that reported previously, as the older literature cites that 75–90% of cases are usually positive.16, 17, 18, 19 However, this finding is similar to that reported by Bobos et al,32 who found that only 15% of cases of Merkel cell carcinoma expressed KIT. The staining we observed was primarily diffuse and weak (Figure 1c) compared to the staining for CD117 observed in GISTs (our positive external control). Our data did not reveal any correlation between KIT expression and clinicopathologic factors (ie, primary vs metastatic disease). Since the reduced number of KIT-positive cases maybe due to differences in scoring (as opposed to actual protein activity or function), we explored the mutational status of this receptor tyrosine kinase in all 32 cases included in this study. Our study did not identify any activating mutations in this receptor, similar to the results of Swick et al,33 who examined nine cases of Merkel cell carcinoma. However, we did observe rare silent mutations in KIT in exon 17 at codon 798. This mutation has been previously reported in one patient with papillary thyroid carcinoma.34 We conclude from our data that in Merkel cell carcinoma, expression of KIT and occurrences of activating mutations in the receptor may be unrelated events. This is true in other tumors as well, such as solid psuedopapillary neoplasms of the pancreas,35 melanomas,36, 37 pulmonary small cell carcinomas38, 39 and breast carcinomas.40
Cytogenetic studies revealed frequent genetic abnormalities in Merkel cell carcinoma,5 but no alterations were observed in chromosome 4q12, the area to which KIT and PDGFRA are mapped.41 In the study of Sihto et al38 of 334 solid tumors, strong KIT protein expression was associated with a mutated KIT gene, with a wild-type KIT gene amplification, or with chromosome 4 aneuploidy. In addition, one of three cases of Merkel cell carcinoma revealed monosomy in chromosome 4 leading to loss of one copy of the KIT gene. This particular case was also negative for KIT.
In the absence of gene rearrangements or mutations in KIT, we examined the expression of the KIT ligand, SCF, to identify if the autocrine/paracrine stimulation of the KIT receptor was a viable alternative pathway of tumor proliferation. SCF expression was weak and diffuse, and did not necessarily correlate with KIT staining. Only 5 of 32 cases (16%) showed coexpression of KIT and SCF. Interestingly, however, autocrine/paracrine stimulation is postulated to be one of the mechanisms by which KIT activation occurs in GISTs,42 small cell lung carcinomas43 and gliomas,44 and autocrine stimulation maybe responsible for KIT activation in these five cases of Merkel cell carcinoma that are KIT+/SCF+.
Although lesional cell staining with the SCF antibody was an uncommon event, we did observe significant staining of neighboring non-lesional cells with SCF. It is possible, therefore, that paracrine effects from exogenous SCF may play a role in activating the KIT receptor. In the absence of activating mutations in KIT, this paracrine stimulation may be the basis for tumor proliferation in Merkel cell carcinoma. Alternatively, downregulation of miRNAs, such as miRNA-221 and miRNA-222 which regulate KIT expression by binding to its 3′-UTR,34, 45, 46 or an unknown mutation in the miRNA recognition site, can also result in receptor expression in the absence of gene amplification or mutations in KIT. This posttranscriptional regulation may also explain the absence of KIT expression in some of our cases of Merkel cell carcinoma, even in the presence of lesional SCF.
In contrast, our analysis of PDGFRA and its ligand, PDGFA, suggests an autocrine mechanism of stimulation for this receptor kinase in the majority of cases of Merkel cell carcinoma. We saw coexpression of PDGFRA and PDGFA in 25 of 31 samples (81%) analyzed. Coexpression of PDGFRA and PDGFA was seen more commonly than coexpression of KIT and SCF (Table 1). Coexpression of PDGFRA and PDGFA has been reported for malignant peripheral nerve sheath tumors and gliomas,26, 44 and an autocrine/paracrine mode of stimulation has been postulated as a mechanism of proliferation. It is possible, therefore, that a similar mode of action may exist for Merkel cell carcinoma. We also found that cases which were negative for KIT receptor expression by immunohistochemistry were positive for PDGFRA, and cases which were likewise negative for PDGFRA were positive for KIT. This finding is similar to that reported for GISTs;21 for example, the signal transduction profiles for GISTs with mutations in PDGFRA, and those with mutations in KIT, were indistinguishable, suggesting that PDGFRA and KIT play similar roles in the oncogenesis of this tumor.23 A similar role may be played by these receptor tyrosine kinases in the pathogenesis of Merkel cell carcinoma.
The clinical efficacy of imatinib mesylate in the treatment of tumors expressing receptor tyrosine kinases appears to be strongly associated with activating mutations in KIT and PDGFRA. We restricted our analysis to the commonly mutated exons of these receptor tyrosine kinases, namely, exons 9, 11, 13 and 17 in KIT and exons 10, 12 and 18 for PDGFRA. We did not identify any known activating mutations in KIT and PDGFRA. However, we identified a mutation in exon 10 of PDGFRA in two cases with Merkel cell carcinoma, which resulted in a serine to proline amino-acid change. These two cases also had another previously unreported silent mutation in the same exon. Interestingly, these two cases represented primary and metastatic samples from the same patient. However, this mutation, which resulted in a non-conservative amino-acid change, has been characterized as an allelic variant in malignant peripheral nerve sheath tumors and gliomas, based on allelic frequencies in normal blood samples.26 It is not clear if this mutation can result in an altered PDGFRA protein which may make these cases of Merkel cell carcinoma responsive to imatinib mesylate.
In the absence of any significant amino-acid change in the most common sites for gain-of-function mutations in these receptor tyrosine kinases, the chance that imatinib mesylate may be an effective therapy in Merkel cell carcinoma is small, unless mutations exist in other exons of KIT or PDGFRA, or in other receptor kinases involved in proliferation pathways. KIT or PDGFRA mutational hot spots may be located in different exons in disparate tumor types. Analysis of entire open reading frames of these receptor tyrosine kinases for activating mutations may be necessary to entirely exclude the possibility that the drug may be effective through the well-known mechanisms of action established for these receptors. In addition, Kimchi-Sarfaty et al47 recently showed that a silent SNP in a mammalian membrane transport protein can affect protein folding, which in turn alters substrate specificity and protein function. The frequent occurrences of silent polymorphisms in receptor tyrosine kinases may therefore be a significant finding, and may very well impart drug sensitivity. It is also interesting that imatinib can inhibit KIT-induced VEGF (vascular endothelial growth factor) expression in a related neoplasm, small cell lung carcinoma.48 This may add another level of action for this drug in the KIT-mediated signaling pathway, thereby increasing the chances of the drug's effectiveness in treating tumors. However, it is not clear from the work of Litz and Krystal48 if the signaling is triggered by a mutant or wild-type version of KIT. In our study of Merkel cell carcinoma, we observed diffuse and weak expression of VEGF in >80% of cases (unpublished data), which is similar to data recently published examining 43 markers on 31 cases of Merkel cell carcinoma via tissue microarray analysis.49 However, the staining we observed for VEGF was independent of KIT expression levels, suggesting that multiple pathways may exist which activate VEGF expression. The data of Fernandez-Figueras et al49 also suggests a likely role for VEGF in tumor progression and the potential benefit of using anticancer treatments targeted against VEGF and other members of its signaling pathway. In this setting, it would be interesting to investigate the role, if any, of imatinib in modulating VEGF expression.
In conclusion, while we found that only a minority of cases of Merkel cell carcinoma had a KIT+/SCF+ dual phenotype, the majority of cases did have a PDGFRA+/PDGFA+ phenotype, suggesting a potential role for this receptor/ligand pair in autocrine/paracrine stimulation of tumor growth and proliferation. In addition, expression of KIT and PDGFRA appear to be related events; for example, we did not find a KIT(−)/PDGFRA(−) immunophenotype. These findings suggest that these two tyrosine kinase receptors have meaningful but redundant functions in the proliferation of this tumor. While we did not find any known activating mutations in KIT or PDGFRA, we were able to find silent mutations, which may potentially lead to an alternate protein function based on changes in protein folding states. Finally, although at present it does not appear that imatinib mesylate has a role to play in the treatment of Merkel cell carcinoma based on known mechanisms of action, our studies do not exclude the possibility of important mutations in other regions of the genes or the role of imatinib in modulating the function of related proteins, such as VEGF. Further studies in these areas may help to elucidate the role of tyrosine kinase inhibitors in the treatment of Merkel cell carcinoma.
References
Agelli M, Clegg LX . Epidemiology of primary Merkel cell carcinoma in the United States. J Am Acad Dermatol 2003;49:832–841.
Goepfert H, Remmler D, Silva E, et al. Merkel cell carcinoma (endocrine carcinoma of the skin) of the head and neck. Arch Otolaryngol 1984;110:707–712.
Allen PJ, Zhang ZF, Coit DG . Surgical management of Merkel cell carcinoma. Ann Surg 1999;229:97–105.
Allen PJ, Bowne WB, Jaques DP, et al. Merkel cell carcinoma: prognosis and treatment of patients from a single institution. J Clin Oncol 2005;23:2300–2309.
Manor E, Bellaiche E, Bodner L . Cytogenetic findings of a primary Merkel cell carcinoma. Cancer Genet Cytogenet 2006;169:78–80.
Toker C . Trabecular carcinoma of the skin. Arch Dermatol 1972;105:107–110.
Tang CK, Toker C . Trabecular carcinoma of the skin: an ultrastructural study. Cancer 1978;42:2311–2321.
Halata Z, Grim M, Bauman KI . Friedrich Sigmund Merkel and his ‘Merkel cell’, morphology, development, and physiology: review and new results. Anat Rec A Discov Mol Cell Evol Biol 2003;271:225–239.
Fernandez-Figueras MT, Puig L, Musulen E, et al. Prognostic significance of p27Kip1, p45Skp2 and Ki67 expression profiles in Merkel cell carcinoma, extracutaneous small cell carcinoma, and cutaneous squamous cell carcinoma. Histopathology 2005;46:614–621.
Heenan PJ, Cole JM, Spagnolo DV . Primary cutaneous neuroendocrine carcinoma (Merkel cell tumor) An adnexal epithelial neoplasm. Am J Dermatopathol 1990;12:7–16.
Pilotti S, Rilke F, Lombardi L . Neuroendocrine (Merkel cell) carcinoma of the skin. Am J Surg Pathol 1982;6:243–254.
Smith KJ, Skelton III HG, Holland TT, et al. Neuroendocrine (Merkel cell) carcinoma with an intraepidermal component. Am J Dermatopathol 1993;15:528–533.
Walsh NM . Primary neuroendocrine (Merkel cell) carcinoma of the skin: morphologic diversity and implications thereof. Hum Pathol 2001;32:680–689.
Sibley RK, Dehner LP, Rosai J . Primary neuroendocrine (Merkel cell?) carcinoma of the skin. I. A clinicopathologic and ultrastructural study of 43 cases. Am J Surg Pathol 1985;9:95–108.
Goessling W, McKee PH, Mayer RJ . Merkel cell carcinoma. J Clin Oncol 2002;20:588–598.
Feinmesser M, Halpern M, Kaganovsky E, et al. c-kit expression in primary and metastatic Merkel cell carcinoma. Am J Dermatopathol 2004;26:458–462.
Llombart B, Monteagudo C, Lopez-Guerrero JA, et al. Clinicopathological and immunohistochemical analysis of 20 cases of Merkel cell carcinoma in search of prognostic markers. Histopathology 2005;46:622–634.
Strong S, Shalders K, Carr R, et al. KIT receptor (CD117) expression in Merkel cell carcinoma. Br J Dermatol 2004;150:384–385.
Su LD, Fullen DR, Lowe L, et al. CD117 (KIT receptor) expression in Merkel cell carcinoma. Am J Dermatopathol 2002;24:289–293.
Pawson T . Regulation and targets of receptor tyrosine kinases. Eur J Cancer 2002;38 (Suppl 5):S3–S10.
Corless CL, Fletcher JA, Heinrich MC . Biology of gastrointestinal stromal tumors. J Clin Oncol 2004;22:3813–3825.
Demetri GD, von Mehren M, Blanke CD, et al. Efficacy and safety of imatinib mesylate in advanced gastrointestinal stromal tumors. N Engl J Med 2002;347:472–480.
Heinrich MC, Corless CL, Demetri GD, et al. Kinase mutations and imatinib response in patients with metastatic gastrointestinal stromal tumor. J Clin Oncol 2003;21:4342–4349.
Fenig E, Nordenberg J, Beery E, et al. Combined effect of aloe-emodin and chemotherapeutic agents on the proliferation of an adherent variant cell line of Merkel cell carcinoma. Oncol Rep 2004;11:213–217.
Yantiss RK, Rosenberg AE, Sarran L, et al. Multiple gastrointestinal stromal tumors in type I neurofibromatosis: a pathologic and molecular study. Mod Pathol 2005;18:475–484.
Holtkamp N, Okuducu AF, Mucha J, et al. Mutation and expression of PDGFRA and KIT in malignant peripheral nerve sheath tumors, and its implications for imatinib sensitivity. Carcinogenesis 2006;27:664–671.
Duensing A, Heinrich MC, Fletcher CD, et al. Biology of gastrointestinal stromal tumors: KIT mutations and beyond. Cancer Invest 2004;22:106–116.
Hartmann C, Xu X, Bartels G, et al. Pdgfr-alpha in 1p/19q LOH oligodendrogliomas. Int J Cancer 2004;112:1081–1082.
Van Gele M, Kaghad M, Leonard JH, et al. Mutation analysis of P73 and TP53 in Merkel cell carcinoma. Br J Cancer 2000;82:823–826.
Liu S, Daa T, Kashima K, et al. The Wnt-signaling pathway is not implicated in tumorigenesis of Merkel cell carcinoma. J Cutan Pathol 2007;34:22–26.
Houben R, Michel B, Vetter-Kauczok CS, et al. Absence of classical MAP kinase pathway signalling in Merkel cell carcinoma. J Invest Dermatol 2006;126:1135–1142.
Bobos M, Hytiroglou P, Kostopoulos I, et al. Immunohistochemical distinction between Merkel cell carcinoma and small cell carcinoma of the lung. Am J Dermatopathol 2006;28:99–104.
Swick BL, Ravdel L, Fitzpatrick JE, et al. Merkel cell carcinoma: evaluation of KIT (CD117) expression and failure to demonstrate activating mutations in the C-KIT proto-oncogene—implications for treatment with imatinib mesylate. J Cutan Pathol 2007;34:324–329.
He H, Jazdzewski K, Li W, et al. The role of microRNA genes in papillary thyroid carcinoma. Proc Natl Acad Sci USA 2005;102:19075–19080.
Cao D, Antonescu C, Wong G, et al. Positive immunohistochemical staining of KIT in solid–pseudopapillary neoplasms of the pancreas is not associated with KIT/PDGFRA mutations. Mod Pathol 2006;19:1157–1163.
Pache M, Glatz K, Bosch D, et al. Sequence analysis and high-throughput immunohistochemical profiling of KIT (CD 117) expression in uveal melanoma using tissue microarrays. Virchows Arch 2003;443:741–744.
Went PT, Dirnhofer S, Bundi M, et al. Prevalence of KIT expression in human tumors. J Clin Oncol 2004;22:4514–4522.
Sihto H, Sarlomo-Rikala M, Tynninen O, et al. KIT and platelet-derived growth factor receptor alpha tyrosine kinase gene mutations and KIT amplifications in human solid tumors. J Clin Oncol 2005;23:49–57.
Tamborini E, Bonadiman L, Negri T, et al. Detection of overexpressed and phosphorylated wild-type kit receptor in surgical specimens of small cell lung cancer. Clin Cancer Res 2004;10:8214–8219.
Simon R, Panussis S, Maurer R, et al. KIT (CD117)-positive breast cancers are infrequent and lack KIT gene mutations. Clin Cancer Res 2004;10:178–183.
Larramendy ML, Koljonen V, Bohling T, et al. Recurrent DNA copy number changes revealed by comparative genomic hybridization in primary Merkel cell carcinomas. Mod Pathol 2004;17:561–567.
Theou-Anton N, Tabone S, Brouty-Boye D, et al. Co expression of SCF and KIT in gastrointestinal stromal tumours (GISTs) suggests an autocrine/paracrine mechanism. Br J Cancer 2006;94:1180–1185.
Hibi K, Takahashi T, Sekido Y, et al. Coexpression of the stem cell factor and the c-kit genes in small-cell lung cancer. Oncogene 1991;6:2291–2296.
Reis RM, Martins A, Ribeiro SA, et al. Molecular characterization of PDGFR-alpha/PDGF-A and c-KIT/SCF in gliosarcomas. Cell Oncol 2005;27:319–326.
Felli N, Fontana L, Pelosi E, et al. MicroRNAs 221 and 222 inhibit normal erythropoiesis and erythroleukemic cell growth via kit receptor down-modulation. Proc Natl Acad Sci USA 2005;102:18081–18086.
Poliseno L, Tuccoli A, Mariani L, et al. MicroRNAs modulate the angiogenic properties of HUVECs. Blood 2006;108:3068–3071.
Kimchi-Sarfaty C, Oh JM, Kim IW, et al. A ‘silent’ polymorphism in the MDR1 gene changes substrate specificity. Science 2007;315:525–528.
Litz J, Krystal GW . Imatinib inhibits c-Kit-induced hypoxia-inducible factor-1alpha activity and vascular endothelial growth factor expression in small cell lung cancer cells. Mol Cancer Ther 2006;5:1415–1422.
Fernandez-Figueras MT, Puig L, Musulen E, et al. Expression profiles associated with aggressive behavior in Merkel cell carcinoma. Mod Pathol 2007;20:90–101.
Acknowledgements
We thank Drs Neeraja Kambham and Erich Schwartz for joint financial support of RK, Lisa Kim and Amarjeet Grewall for laboratory assistance and Anet James for assistance with the graphics.
Author information
Authors and Affiliations
Corresponding author
Additional information
Disclosure/conflict of interest
The authors state no conflict of interest.
Rights and permissions
About this article
Cite this article
Kartha, R., Sundram, U. Silent mutations in KIT and PDGFRA and coexpression of receptors with SCF and PDGFA in Merkel cell carcinoma: implications for tyrosine kinase-based tumorigenesis. Mod Pathol 21, 96–104 (2008). https://doi.org/10.1038/modpathol.3800980
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.3800980
Keywords
This article is cited by
-
The biology and treatment of Merkel cell carcinoma: current understanding and research priorities
Nature Reviews Clinical Oncology (2018)
-
Platelet-derived growth factor may be a potential diagnostic and prognostic marker for cholangiocarcinoma
Tumor Biology (2012)
-
High KIT and PDGFRA are associated with shorter patients survival in gastroenteropancreatic neuroendocrine tumors, but mutations are a rare event
Journal of Cancer Research and Clinical Oncology (2012)
