
Abstract
Despite its alarming appearance, spermatocytic seminoma virtually never metastasizes. We hypothesized that this paradox may at least be partially related to increased apoptosis compared to metastasizing germ cell tumors since high expression of proapoptotic factors correlates with indolent behavior in other tumor systems, notably CD30-positive cutaneous lymphoma, another neoplasm where phenotype and behavior do not match. We therefore compared apoptosis and apoptotic regulators in 17 spermatocytic seminomas (2 with sarcoma) and 18 usual seminomas by light microscopy and using immunostains for caspase-3, p53, bcl-2, bcl-xL, FADD, FAS and survivin. We found significantly greater numbers of apoptotic cells and activated caspase-3-positive cells in spermatocytic seminoma compared to usual seminoma (P<0.01). There was over a 10-fold range in apoptotic cells in usual seminoma but only a 4-fold variation in spermatocytic seminoma. Spermatocytic seminoma had decreased p53 expression compared to usual seminoma, with marked variation in bcl-2 expression and increased FADD. The two sarcomas in spermatocytic seminoma, however, showed decreased apoptosis and caspase-3 reactivity, with upregulation of p53 and bcl-2 and decreased FADD expression. We conclude that apoptosis, caspase-3 and FADD expression are increased in spermatocytic seminoma compared to usual seminoma. Apoptotic parameters are decreased in sarcomatous transformation of spermatocytic seminoma. The increased apoptosis of spermatocytic seminoma, possibly mediated by FAS independent activation of the death receptor pathway, may provide some insight into its excellent prognosis. The variation in apoptosis of usual seminomas merits investigation as a prognostic parameter.
Similar content being viewed by others

Main
Spermatocytic seminoma is an uncommon testicular neoplasm with distinctive clinicopathologic features.1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12 It occurs in older men, never arises in extratesticular sites, and, with extremely rare possible exceptions,13 exhibits a benign course. Spermatocytic seminoma does not occur with other germ cell tumors, but it is rarely associated with high-grade sarcoma.14, 15 Unlike other testicular tumors, it does not appear to be linked to cryptorchidism,3, 16 is not associated with serum marker elevation,3, 17 and probably does not share epidemiologic features with the usual forms of testicular germ cell tumors.
Microscopically, the most distinctive feature is its cellular polymorphism, represented by three cell types (small, intermediate-sized and large) typically arranged in sheets punctuated by occasional cystic spaces filled with eosinophilic fluid. Rare ‘anaplastic’ variants lack the polymorphism and consist of a relatively monomorphic population of intermediate-sized cells with prominent nucleoli, with foci of typical spermatocytic seminoma elsewhere.18 The mitotic rate is often high and extra-testicular and vascular invasion may be seen.
The benign clinical course of spermatocytic seminoma (unless complicated by sarcoma) is therefore unexpected given its morphologic features. An additional striking microscopic finding that we have noted is a very prominent number of apoptotic tumor cells; we therefore, hypothesized that the benign biologic behavior of spermatocytic seminoma may at least be partially related to increased apoptosis compared to metastasizing germ cell tumors. We therefore compared apoptosis and apoptotic regulators in spermatocytic and usual seminomas.
Materials and methods
The consultation and routine files of Indiana University and Methodist Hospitals were searched for orchiectomy specimens of spermatocytic seminoma and usual seminoma. Seventeen spermatocytic seminomas and 18 randomly selected usual seminomas were retrieved. Two of the spermatocytic seminomas also had a sarcoma component. Either unstained sections or paraffin blocks were available for study. All histologic and immunohistochemical analyses were performed on 3–4 μm thick sections from formalin-fixed, paraffin-embedded tissue. None of the patients received chemotherapy or radiotherapy before orchiectomy. Mitotic rates were determined by counting 10 random high-power fields (HPF) (× 40 objective) using an Olympus BX40 microscope equipped with × 10 WH/22 eyepieces (field area=0.25 mm2).
Evaluation of Apoptosis
Apoptotic cells were quantified by light microscopy on hematoxylin and eosin (H&E)-stained sections by averaging the number of cells with homogeneously dense chromatin or karyorrhectic nuclear fragments in photographs of five randomly selected × 40 fields. The cases were evaluated by two independent examiners (EFB and TMU). Only unequivocal apoptotic bodies were counted.
Immunohistochemistry
Immunohistochemical stains for p53 and bcl-2 were performed using an automated immunostainer (Dako Cytomation, Carpinteria, CA, USA). The antibody clones and staining conditions are listed in Table 1. Survivin, bcl-xL, p-FADD, FAS and activated caspase-3 stains were performed manually. Briefly, endogenous peroxidase activity was blocked with 3% hydrogen peroxide in methanol, and endogenous biotin activity was blocked using a protein block. After antigen retrieval (if necessary), the slides were incubated with primary antibody. The binding was detected with a secondary antibody tagged with horseradish peroxidase. The peroxidase activity was developed with 3,3-diaminobenzidene and counterstained with hematoxylin. Control slides and appropriate internal positive controls were reviewed and determined adequate in all cases. The slides were scored on the basis of nuclear staining for p53 and FADD and cytoplasmic expression for survivin, bcl-2, bcl-xL, Fas and activated caspase-3 in tumor cells. Counts of individually staining cells or estimates of percent of positive cells were performed.
Statistical Analysis
The significance of differences in the proportions of apoptotic cells and cells expressing immunohistochemical markers in spermatocytic seminoma and usual seminoma was determined by the t-test procedure as performed through a statistical analysis website: (http://www.physics.csbsju.edu/stats/t-test.html). Correlation coefficients were calculated by utilizing Microsoft Excel spreadsheet statistical function.
Results
Clinical Information
The 17 spermatocytic seminoma patients ranged from 32 to 77 years old (mean, 50 years). The majority of tumors were in the right testis (n=12). In two patients (38 and 39 years old), the spermatocytic seminoma had a sarcomatous component. Patients with usual seminoma ranged from 20 to 57 years old (mean, 37 years).
Microscopic Features
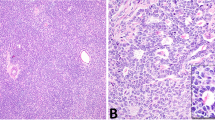
The spermatocytic seminomas had nests of tumor cells, sometimes with cystic change, with sparse intervening stroma. Usually, the typical ‘tripartite’ cell population was identified (Figure 1), although two cases were relatively monomorphic (‘anaplastic’ spermatocytic seminoma18). Two additional cases showed a combination of spermatocytic seminoma and sarcoma. The first showed nests of spermatocytic seminoma that were surrounded and infiltrated by sarcoma comprised of sheets of plump, pleomorphic cells (Figure 2) alternating with fascicles of spindle cells. The predominant cell type had enlarged, irregularly shaped, overlapping nuclei, vesicular chromatin, prominent single nucleoli and scant-to-moderate amounts of eosinophilic cytoplasm with indistinct cellular borders. Numerous bizarre tumor giant cells were present. A fairly distinct interface between spermatocytic seminoma and sarcoma existed in the other case, with the sarcoma consisting of interlacing fascicles of undifferentiated spindle cells (Figure 3). Tumor giant cells were a focal feature. Frequent mitotic figures and geographic necrosis were evident in both cases. The spermatocytic seminomas had 6–101 mitotic figures per 10 HPF (mean, 44). This compared with 9–55 mitotic figures per 10 HPF (mean, 28) in the usual seminomas.

Spermatocytic seminoma showing numerous apoptotic cells amidst variably sized tumor cells.

Sarcoma in spermatocytic seminoma, showing pleomorphic, plump spindle cells and bizarre giant cells.

Sarcoma in spermatocytic seminoma showing: (a) an ill-defined fascicular arrangement of (b) spindle cells with frequent mitotic figures.
Evaluation of Apoptosis by Light Microscopy
Apoptotic bodies were counted in 13 pure spermatocytic seminomas, two spermatocytic seminomas with sarcoma, and 13 ‘pure’ usual seminomas (Table 2). Spermatocytic seminomas demonstrated significantly increased apoptotic cells (Figure 1), with an average of 96 per HPF (range, 51–183/HPF), compared to usual seminoma (mean, 32/HPF; range, 7–78/HPF) (P<0.01). Paired analysis of apoptotic and mitotic rates in the spermatocytic seminomas (excluding the two sarcomas) showed a weak, positive correlation (r=0.29). Apoptosis was decreased (mean, 30/HPF; 17 and 42/HPF) in the sarcoma of the two cases with this component. Unexpectedly, a few usual seminomas showed high apoptotic counts (40–78/HPF) comparable to or exceeding those of several of the spermatocytic seminomas. Paired analysis of apoptotic and mitotic rates in the usual seminomas demonstrated a positive correlation (r=0.54).
Immunohistochemistry
Caspase-3
The spermatocytic seminomas showed increased expression of activated caspase-3 (mean, 18 positive cells/HPF) (Figure 4a) compared to usual seminoma (mean, 9 positive cells/HPF) (Figure 4b; P<0.01). Caspase-3 expression was relatively low in the two sarcomas (mean, 3 positive cells/HPF) (Figure 4c; Table 2).

Caspase-3 expression in (a) spermatocytic seminoma, (b) usual seminoma and (c) sarcoma component of spermatocytic seminoma.
Phospho-FADD (p-FADD)
There was increased expression of p-FADD, showing granular staining in both nuclei and cytoplasm, in spermatocytic seminoma compared to usual seminoma. In 12 cases of spermatocytic seminoma, an average of 71% of cells expressed p-FADD (range 40–95%) (Figure 5a), whereas usual seminomas had an average of 46% of positively staining tumor cells (range 30–60%) (Figure 5b) (P<0.01); 3 cases of spermatocytic seminoma were excluded because of technical problems with the stain. Decreased p-FADD-reactivity was noted in the two cases of sarcoma (20 and 15% positive cells, respectively) (Figure 5c).

Expression of p-FADD in (a) spermatocytic seminoma, (b) usual seminoma and (c) sarcoma component of spermatocytic seminoma.
Bcl-2
Spermatocytic seminomas averaged 12% positive tumor cells for bcl-2 protein (range 0–70), with expression in 14 of 16 (87.5%) cases (Figure 6a). No bcl-2 reactivity was noted in the tumor cells of 18 usual seminomas. The tumor cells in the two sarcomas showed markedly increased bcl-2 expression (95 and 80%) (Figure 6b).

Expression of bcl-2 in (a) spermatocytic seminoma and (b) sarcoma component of spermatocytic seminoma.
p53
Spermatocytic seminomas were mostly negative for p53 expression (<1%) of tumor cells; range, 0–5%), whereas usual seminomas showed widely scattered positively staining tumor cells (mean, 7%). Increased expression of p53 protein was noted in the two sarcomas, where 60 and 90% of cells were positive.
FAS
There was little expression of FAS in both spermatocytic seminomas and usual seminomas (1 and <1%, respectively). Ten percent of tumor cells showed positivity for FAS in one of the two sarcoma cases, whereas no staining was noted in the other case.
Bcl-xL and survivin
None of the cases showed any staining for bcl-xL or survivin.
Discussion
Apoptosis or programmed cell death is a distinct, intrinsic cell mechanism that occurs in various physiological and pathological situations.19 In an average human adult, 50–70 billion cells a day undergo apoptosis,20 and inability to undergo apoptosis is implicated in the development of cancer.
Apoptosis is induced by a cascade of molecular events that may be initiated in distinct ways and culminate in the activation of caspases.21 The process of apoptosis may be divided into an initiation phase, during which caspases become catalytically active, and an execution phase, during which these enzymes act to cause cell death. Initiation of apoptosis occurs principally by signals from two distinct but convergent pathways—the extrinsic, or receptor initiated, pathway and the intrinsic, or mitochondrial, pathway. Both pathways converge to activate caspases.
The extrinsic (death receptor initiated) pathway is initiated by engagement of cell surface death receptors on a variety of cells.22 Death receptors are members of the tumor necrosis factor (TNF) receptor family that contain a cytoplasmic domain involved in protein–protein interactions that is called the death domain because it is essential for delivering apoptotic signals. Upon binding of Fas antigen (CD95) to its ligand (FasL or CD95L), the Fas receptor activates a 26-kDa protein known as FADD (Fas-associated with death domain).20 Besides the FasL–Fas complex, FADD protein may interact with other ligand–receptor complexes, including TNF-α/TNFR-1 and 2; APO-3L/TWEAK/DR-3, APC-3; and APO-2L/TRAIL/DR4-5.23 This connection, in turn, initiates a cascade of caspases, a group of ubiquitous cysteine proteases that are activated by proteolytic cleavage.24, 25 Beginning with activation of caspase-8, this cascade culminates in the activation of caspase-3, a protein that destroys key cell structural proteins, such as fodrin, lamin A and poly(ADP-ribose) polymerase to produce fragmentation of the nucleus and cytoplasm.26
The intrinsic (mitochondrial) pathway is the result of increased mitochondrial permeability and release of proapoptotic molecules into the cytoplasm, without a role for death receptors.27, 28 Growth factors and other survival signals stimulate the production of antiapoptotic members of the bcl-2 family of proteins.29 Members of the bcl-2 family proteins play a pivotal role in the regulation of the mitochondrial pathway, since these proteins localize to intracellular membranes.30 They comprise both antiapoptotic members—for example, bcl-2, bcl-xL and mcl-1—as well as proapoptotic molecules such as bax, bek, bad and BH3 domain-only molecules which link the death receptor pathway to the mitochondrial pathway (bid, bim, puma and noxa). Upon apoptosis induction, proapoptotic bcl-2 proteins with multidomains such as bax translocate from the cytoplasm to the outer mitochondrial membrane, where they form a pore-like structure, thereby promoting cytochrome c release.31 This translocation to mitochondria can be triggered by bcl-2 proteins, which have a BH3 domain only. Bcl-2 or bcl-xL exert their antiapoptotic functions, at least in part, by sequestering BH3 domain-only proteins in stable complexes, thereby preventing activation and translocation of bax or bak to mitochondria. In addition, bcl-2 and bcl-xL block apoptosis by preventing cytochrome c release through a direct effect on mitochondrial channels.32, 33, 34
Tumor suppressor protein p53 accumulates when DNA is damaged and arrests the cell cycle to allow additional time for repair.35 However, if the repair process fails, p53 triggers apoptosis by inducing the activation of apoptosis-inducing genes. Bax and IGF–BP3 are the two p53-responsive genes that carry the death commands of p53. Loss of p53 or its inactivation by mutation is the most common genetic alteration in human tumors. Overexpression of mutated p53 protein by immunohistochemistry is seen in many human cancers.
Death by apoptosis is a normal phenomenon that serves to eliminate cells that are no longer needed. Activation of apoptosis pathways is also a key mechanism by which cytotoxic drugs kill tumor cells.32 It causes cell death in tumors, most frequently during regression but also in actively growing tumors. Although rapidly proliferating, high-grade tumors are characterized by increased cell death rates, other tumors expressing high degrees of apoptosis are sometimes characterized by indolent behavior and occasional spontaneous regression. Clarke et al26 provided strong evidence that death-receptor-mediated apoptosis may be responsible for the greater frequency of spontaneous regression observed in primary cutaneous CD30-positive lymphoproliferative disorders as compared with systemic anaplastic large cell lymphoma. Kikuchi and Nishikawa36 demonstrated that CD30-positive lymphoproliferative disorders of the skin, which characteristically have an indolent course, undergo significantly more apoptosis than other cutaneous lymphoproliferative disorders. The demonstration that many CD30-positive neoplastic cells strongly express the FAS receptor,37, 38 a molecule known to regulate a key apoptotic pathway, as well as FADD and caspase-3, provided further support for involvement of apoptosis in spontaneous regression of these cutaneous lymphomas.26
Along these lines, we hypothesized that apoptosis may play a role in the indolent behavior of spermatocytic seminomas compared to usual seminomas. We do not wish to imply, however, that high levels of apoptosis alone provide a satisfactory explanation for tumor indolence, since it is very clear that many tumors with high apoptotic rates disseminate widely and are lethal. Although they both derive from germ cells of seminiferous tubules, spermatocytic seminoma and usual seminoma differ markedly in biologic behavior. Understanding what causes spermatocytic seminoma to remain localized to the testis may provide insight about intrinsic mechanisms of tumor progression or regression. In this study, we compared the degree of apoptosis in spermatocytic seminomas and usual seminomas, as expressed by numbers of apoptotic bodies by light microscopy, as well as by expression of death receptors, adaptor proteins and initiator caspases by immunohistochemistry.
We found that apoptotic counts, FADD and activated caspase-3 expression are increased 2- to 3-fold in spermatocytic seminomas compared to usual seminomas. These findings indicate higher rates of apoptosis in spermatocytic seminoma through activation of the extrinsic apoptotic pathway. Surprisingly, FAS receptors were not overexpressed in spermatocytic seminomas, suggesting FAS-independent activation of the death receptor pathway. These data are discordant with a previous study by Kersemaekers et al39 in which spermatocytic seminomas were the only germ cell tumors that expressed both Fas and FasL. In contrast to spermatocytic seminoma, apoptotic parameters were decreased in sarcomatous transformation of spermatocytic seminoma, in keeping with the highly aggressive course of those neoplasms.
Successful metastasis depends upon a number of tumor activities, including stromal invasion, initial vascular intrusion, maintenance of cell viability during intravascular migration, subsequent extravascular invasion and successful growth of tumor cells in a secondary environment through parasitization of essential nutrients there. Although spermatocytic seminoma demonstrates high degrees of apoptosis, it is unclear if increased apoptosis in spermatocytic seminoma contributes to its lack of metastatic behavior. While spermatocytic seminoma cells are capable of invading vessels, they may be vulnerable to immunologic destruction while in the circulation, unable to exit vascular spaces, or lack the ability to obtain essential growth factors at extra-testicular sites. Additional studies are needed to explore further the role that apoptosis may play in any or all of these processes. It certainly seems plausible that a tumor with high apoptotic activity may be vulnerable to destruction during one or more of these steps. The correlation previously mentioned concerning apoptosis and the indolent behavior of CD30-positive cutaneous lymphomas26, 36 lends some credence to a role for apoptosis in reducing tumor virulence. Nonetheless, the role of high apoptotic rates in contributing to tumor behavior in spermatocytic seminomas remains speculative at our current state of knowledge.
The antiapoptotic proteins, bcl-xL and survivin, were not expressed in any of the lesions studied, including sarcomas and usual seminomas. Expression of bcl-2 in spermatocytic seminoma was intermediate between that of usual seminoma (which showed no positivity) and sarcomas (which overexpressed bcl-2). Variable prognostic results have been found in a number of studies that looked at bcl-2 and bax expression in a variety of neoplasms.40, 41, 42, 43, 44, 45 Further studies using antibodies to individual members of bcl-2 family proteins are needed to determine the role of these proteins in the prognosis of germ cell tumors of the testis. Comparison of bcl-2 expression in non-neoplastic testis and testicular tumors might also be helpful.
p53 protein was overexpressed in sarcomas, weakly expressed in usual seminomas and virtually nonexistent in spermatocytic seminomas. Although it is usually mutated p53 that is detected by immunohistochemical methods,46 this does not appear to be the case for p53 overexpression in germ cell tumors.47 The role of p53, if any, in the improved prognosis of spermatocytic seminoma compared to usual seminoma, therefore, remains unclear. Its marked overexpression in the sarcomatous transformation of spermatocytic seminoma likely does reflect mutation and correlates with the aggressive behavior of these tumors, as has been shown in other neoplasms.48, 49
Interestingly, 7 of 13 cases of usual seminoma demonstrated quite high numbers of apoptotic bodies (range, 32–78/HPF), whereas in the remainder the apoptotic rate was low. We attempted to correlate other features of possible prognostic significance in usual seminoma with apoptotic rates by comparing the extent of lymphocytic infiltration, granulomatous reaction and tumor size of the usual seminomas having the lowest and highest numbers of apoptotic tumor cells (data not shown). There was no apparent relationship of these features with apoptotic rate in our sample of usual seminomas. We speculate, however, that those usual seminomas with high apoptotic rates might have a more favorable prognosis, perhaps allowing for conservative management after orchiectomy. This may be a fruitful area for further study, even though not the focus of the current investigation.
References
Cummings OW, Ulbright TM, Eble JN, et al. Spermatocytic seminoma: an immunohistochemical study. Hum Pathol 1994;25:54–59.
Rosai J, Silber I, Khodadoust K . Spermatocytic seminoma I. Clinicopathologic study of six cases and review of the literature. Cancer 1969;24:92–102.
Talerman A . Spermatocytic seminoma. Clinicopathologic study of 22 cases. Cancer 1980;45:2169–2176.
Scully RE . Spermatocytic seminoma of the testis. A report of 3 cases and review of the literature. Cancer 1961;14:788–794.
Chung PWM, Bayley AJS, Sweet J, et al. Spermatocytic seminoma: a review. Eur Urol 2004;45:495–498.
Ulbright TM . Testicular and paratesticular tumors. In: Mills SE, Carter D, Greenson JK, Oberman HA, Reuter VE, Stoler MH (eds). Sternberg's Diagnostic Surgical Pathology, 3rd edn. Lippincott Williams & Wilkins: Philadelphia, 2004, pp 2167–2232.
Woodward PJ, Mostofi FK, Talerman A, et al. Germ cell tumors. In: Eble JN, Sauter G, Epstein JI, Sesterhenn IA (eds). Pathology and Genetics of Tumours of the Urinary System and Male Genital Organs. IARC Press: Lyon, 2004, pp 221–249.
Masson P . Étude sur le seminome. Rev Can Biol 1946;5:361–387.
Damjanov I . Pathology of testicular tumors. In: Raghavan D, Scher HI, Leibel SA, Lange PH (eds). Principles and Practice of Genitourinary Oncology. Lippincott-Raven: Philadelphia, 1997, pp 653–662.
Pendlebury S, Horwich A, Dearnaley DP, et al. Spermatocytic seminoma: clinicopathological review of ten patients. Clin Oncol (R Coll Radiol) 1996;8:316–318.
Thomas G, Jones W, VanOosterom A, et al. Consensus statement on the investigation and management of testicular seminoma 1989. Prog Clin Biol Res 1990;357:285–294.
Ulbright TM . Germ cell neoplasms of the testis. Am J Surg Pathol 1993;17:1075–1091.
Matoska J, Ondrus D, Hornak M . Metastatic spermatocytic seminoma. A case report with light microscopic, ultrastructural and immunohistochemical findings. Cancer 1988;62:1197–1201.
True LD, Otis CN, Delprado W, et al. Spermatocytic seminoma of testis with sarcomatous transformation. A report of 5 cases. Am J Surg Pathol 1988;12:75–82.
Floyd CF, Ayala AG, Logothetis J, et al. Spermatocytic seminoma with associated sarcoma of the testis. Cancer 1988;61:409–414.
Eble JN . Spermatocytic seminoma. Hum Pathol 1994;25:1035–1042.
Burke AP, Mostofi FK . Spermatocytic seminoma: a clinicopathologic study of 79 cases. J Urol Pathol 1993;1:21–32.
Albores-Saavedra J, Huffman H, Alvarado-Cabrero I, et al. Anaplastic variant of spermatocytic seminoma. Hum Pathol 1996;27:650–655.
Hengartner MO . The biochemistry of apoptosis. Nature 2000;407:777–783.
Delhalle S, Duvoix A, Schnekenburger M, et al. An introduction to the molecular mechanisms of apoptosis. Ann NY Acad Sci 2003;1010:1–8.
Kumar V, Abbas AK, Fausto N . Cellular adaptations, cell injury, and cell death. In: Kumar V, Abbas AK, Fausto N (eds). Robbins and Cotran: Pathologic Basis of Disease, 7th edn. Elsevier Saunders: Philadelphia, 2005, pp 3–46.
Wallach D, Varfolomeev EE, Malinin NL, et al. Tumor necrosis factor receptor and Fas signaling mechanisms. Annu Rev Immunol 1999;17:331.
Wajant H . The Fas signaling pathway: more than a paradigm. Science 2002;296:1635–1636.
Dempsey PW, Doyle SE, He JQ, et al. The signaling adaptors and pathways activated by TNF superfamily. Cytokine Growth Factor Rev 2003;14:193–209.
Green DR . Apoptotic pathways: paper wraps stone blunts scissors. Cell 2000;102:1–4.
Clarke LE, Bayerl MG, Bruggeman BS, et al. Death receptor apoptosis signaling mediated by FADD in CD30-positive lymphoproliferative disorders involving the skin. Am J Surg Pathol 2005;29:452–459.
Scorrano L, Korsmeyer SJ . Mechanisms of cytochrome release by proapoptotic BCL-2 family members. Biochem Biophys Res Commun 2003;304:437–444.
Ravagnan L, Roumier T, Kroemer G . Mitochondria, the killer organelles and their weapons. J Cell Physiol 2002;192:131.
Adams JM, Cory S . The Bcl-2 protein family: arbiters of cell survival. Science 1998;281:1322–1326.
Antonsson B, Martinou JC . The Bcl-2 protein family. Exp Cell Res 2000;256:50–57.
Cheng EH, Wei MC, Weiler S, et al. Bcl-2, Bcl-xL sequester BH3 domain-only molecules preventing Bax- and Bak-mediated mitochondrial apoptosis. Mol Cell 2001;8:705–711.
Debatin KM . Apoptosis pathways in cancer and cancer therapy. Cancer Immunol Immunother 2004;53:153–159.
Schulze-Osthoff K, Ferrari D, Los M, et al. Apoptosis signaling by death receptors. Eur J Biochem 1998;4:439–459.
Cory S, Adams JM . The Bcl2 family: regulators of the cellular life-or-death switch. Nature Rev Cancer 2002;2:647.
Kumar V, Abbas AK, Fausto N . Neoplasia. In: Kumar V, Abbas AK, Fausto N (eds). Robbins and Cotran: Pathologic Basis of Disease, 7th edn. Elsevier Saunders: Philadelphia, 2005, pp 269–342.
Kikuchi A, Nishikawa T . Apoptotic and proliferating cells in cutaneous lymphoproliferative disorders. Arch Dermatol 1997;133:829–833.
Mori M, Manuelli C, Pimpinelli N, et al. CD30-CD30 ligand interaction in primary cutaneous CD30(+) T-cell lymphomas: a clue to the pathophysiology of clinical regression. Blood 1999;94:3077–3083.
Paulli M, Berti E, Boveri E, et al. Cutaneous CD30(+) lymphoproliferative disorders: expression of bcl-2 and proteins of the tumor necrosis factor receptor superfamily. Hum Pathol 1998;29:1223–1230.
Kersemaekers AMF, van Weeren PC, Oosterhuis JW, et al. Involvement of the Fas/FasL pathway in the pathogenesis of germ cell tumors of the adult testis. J Pathol 2002;4:423–429.
Bargou RC, Daniel PT, Mapara MY, et al. Expression of the bcl-2 gene family in normal and malignant breast tissue: low bax-alpha expression in tumor cells correlates with resistance towards apoptosis. Int J Cancer 1995;60:854–859.
Prokop A, Wieder T, Sturm I, et al. Relapse in childhood acute lymphoblastic leukemia is associated with a decrease of the Bax/Bcl-2 ratio and loss of spontaneous caspase-3 processing in vivo. Leukemia 2000;14:1606–1613.
Sturm I, Kohne CH, Wolff G, et al. Analysis of the p53/Bax pathway in colorectal cancer: low Bax is a negative prognostic factor in patients with resected liver metastases. J Clin Oncol 1999;17:1364–1374.
Leek RD, Kaklamanis L, Pezzella F, et al. Bcl-2 in normal human breast and carcinoma, association with oestrogen receptor-positive, epidermal growth factor receptor-negative tumours and in situ cancer. Br J Cancer 1994;69:135–139.
Pezzella F, Turley H, Kuzu I, et al. Bcl-2 protein in non-small cell lung carcinoma. New Engl J Med 1993;329:690–694.
Nunez G, London L, Hockenbery D, et al. Deregulated Bcl-2 gene expression selectively prolongs survival of growth factor-deprived hematopoietic cell lines. J Immunol 1990;144:3602–3610.
Ogretmen B, Safa AR . Expression of the mutated p53 tumor suppressor protein and its molecular and biochemical characterization in multidrug resistant MCF-7/Adr human breast cancer cells. Oncogene 1997;14:499–506.
Fleischhacker M, Strohmeyer T, Imai Y, et al. Mutations of the p53 gene are not detectable in human testicular germ cell tumors. Mod Pathol 1994;7:435–439.
Tortola S, Marcuello E, Gonzalez I, et al. p53 and K-ras gene mutations correlate with tumor aggressiveness but are not of routine prognostic value in colorectal cancer. J Clin Oncol 1999;17:1375–1381.
Ahnen DJ, Feigl P, Quan G, et al. Ki-ras mutation and p53 overexpression predict the clinical behavior of colorectal cancer. A Southwest oncology group study. Cancer Res 1998;58:1149–1158.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Bishop, E., Badve, S., Morimiya, A. et al. Apoptosis in spermatocytic and usual seminomas: a light microscopic and immunohistochemical study. Mod Pathol 20, 1036–1044 (2007). https://doi.org/10.1038/modpathol.3800933
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.3800933
