
Abstract
We report detailed clinical and pathologic features of four cases of anaplastic lymphoma kinase-positive diffuse large B-cell lymphoma (ALK-DLBCL), a rare entity with only 29 currently reported cases. This study is the third largest of all reported series. Biopsies from four adult patients aged 41, 49, 53, and 71 years (three lymph nodes and one nasopharyngeal mass) exhibited immunoblastic/plasmablastic morphology. By immunohistochemistry and/or flow cytometry, they expressed cytoplasmic ALK-1, CD138, VS38 (3/3), monoclonal cytoplasmic light chain, CD45, EMA, CD4, and CD57 (2/3), and were negative for CD3, CD30, CD56, and TIA-1. Two showed variable CD79a expression, and one had rare CD20(+) cells. Two of three cases exhibited rare CD43(+) reactivity. One case showed scattered cytokeratin(+) cells, which could possibly lead to a misdiagnosis of carcinoma. After CHOP and radiotherapy, two stage I patients were free of disease at 58 and 36 months, whereas a stage IV patient was dead of disease at 22 months.
Similar content being viewed by others

Main
In 1997, seven cases of a new subtype of diffuse large B-cell lymphoma expressing the anaplastic lymphoma kinase gene product (ALK-DLBCL) were reported by Delsol et al.1 This lymphoma was identified due to its characteristic lack of CD30 expression in an otherwise large series of classical T-/null cell ALK-positive anaplastic large cell lymphomas (ALCL).1 ALK-DLBCL exhibits an immunoblastic/plasmablastic morphology, often an intrasinusoidal growth pattern, and is derived from B cells based on expression of monotypic light chain. It shows an unique immunophenotypic profile characterized by a lack of B (CD20, CD79a)- and T-lineage (CD3) markers and CD30, but expression of VS38 and CD138 (plasmacytic markers), variable expression of CD4 and CD57, and cytoplasmic, granular ALK reactivity.
Initially, molecular and protein analyses failed to reveal an ALK gene rearrangement.1 Recently however, Clathrin/ALK (CLTC/ALK) and NPM/ALK gene rearrangements have been identified in 16 and three cases of ALK-DLBCL, respectively.2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13 All of these cases display the morphologic and immunophenotypic features of ALK-DLBCL, as originally described by Delsol et al. Importantly, ALK positivity and ALK gene rearrangements can be seen in B-cell lymphomas and are now no longer uniquely restricted to the T-cell lymphoma, ALCL.
To our knowledge, only 29 cases of ALK-DLBCL have been reported thus far in the literature. Herein, we report detailed clinical and pathologic features of four new adult cases of this rare entity.
Materials and methods
Four total cases of ALK-positive DLBCL were identified: three from the hematology consultation files at the University of Texas Southwestern Medical Center from 1997 to 2002 and one from University of New Mexico, 2006. One case was originally diagnosed as an anaplastic plasmacytoma, but was subsequently reclassified. Clinical and laboratory information for each of the four patients was obtained through physician interview. The mean disease-free survival for low stage patients (alive with disease) and the overall median survival for high stage patients (dead of disease) were calculated from the data reported in the literature.
Routine hematoxylin and eosin-stained sections were prepared from formalin-fixed and/or B5-fixed paraffin blocks. Immunohistochemical analysis included a broad panel of antibodies (Table 1). Fresh tissue from case 2 was submitted for flow cytometric analysis. The cell preparation and data analysis were performed as previously described.14
Fluorescence in situ hybridization (FISH) for the Clathrin/ALK fusion associated with t(2;17)(p23;q23) was performed on case 3 as previously described.2 FISH evaluation for an ALK gene rearrangement using the ALK break apart probe from Vysis, Inc. (Downers Grove, IL, USA) was performed on case 4 on a paraffin-embedded tissue section.
Genetic studies could not be attempted on the other two cases owing to lack of available remaining specimen.
Results
Case Reports
A summary of the clinical features of the four patients is provided in Table 2. All patients were adults; two female and two male.
Case 1
A 41-year-old female presented with left cervical lymphadenopathy (2.8 × 1.8 × 1.0 cm). Hematologic studies, lactate dehydrogenase, and serum protein electrophoresis were all within normal limits. A computed tomography (CT) scan of the thorax and abdomen showed no mass lesions or additional lymphadenopathy. An excisional biopsy of the lymph node was performed. A staging bone marrow was negative for involvement by lymphoma. The patient underwent six cycles of CHOP followed by left neck irradiation for stage 1 disease. Follow-up imaging studies show no evidence of lymphoma. At 58 months, she was alive without disease.
Case 2
A 49-year-old female presented with an enlarged left cervical lymph node that she had noticed for about 1 month. She was otherwise asymptomatic and had no significant past medical history. Hematologic and liver function studies were within normal limits. A CT scan of the neck revealed left submandibular lymphadenopathy. CT scans of the head, chest, pelvis, and abdomen were unremarkable. Biopsy of the left cervical lymph node was performed. Staging bone marrow was negative for involvement by lymphoma. She received four cycles of CHOP followed by involved field irradiation for stage 1 disease. At 36 months, she was alive and free of disease.
Case 3
A 71-year-old male presented with a chief complaint of hemoptysis for six weeks. Physical exam revealed a large nasopharyngeal mass (approximately 5.5 cm) without peripheral lymphadenopathy. CBC showed mild normochromic, normocytic anemia. CT scan demonstrated abdominal and retroperitoneal lymphadenopathy. A biopsy of the nasopharyngeal mass was performed. A staging bone marrow showed low level involvement by chronic lymphocytic leukemia, but no evidence of large cell lymphoma. The patient received CHOP and nasopharyngeal irradiation for stage IV disease. Follow-up imaging at 6 months revealed residual pelvic disease. Twenty-two months from diagnosis, the patient died of disease.
Case 4
A 53-year-old male presented in Mexico with a rapidly enlarging left neck mass. A biopsy of the mass was performed in Mexico. The patient subsequently sought medical attention in the Southwestern United States. By physical examination and radiologic studies, no additional sites of disease are currently identified. A bone marrow biopsy was negative.
Morphology
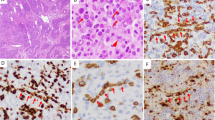
All four cases show similar morphologic features (Figures 1 and 2). All cases showed diffuse effacement of the normal architecture by sheets of tumor cells. Sinusoidal infiltration was seen in case 2 (Figure 1a) along with focal coagulative necrosis. Case 1 exhibited prominent, ectatic, blood-filled vascular spaces. Case 3 exhibited a ‘starry-sky’ pattern. Case 4 shows a prominent intravascular component. The neoplastic cells in all cases were large with round, regular, centrally located nuclei, dispersed chromatin, a single, central, prominent nucleolus, and moderate eosinophilic or amphophilic cytoplasm. Occasional binucleate or rare multinucleate cells resembling Reed–Sternberg cells were seen.

Architectural and cytologic features of ALK-DLBCL. Diffuse proliferation of large cells with round, regular nuclei; single, central, eosinophilic nucleoli, dispersed chromatin, and moderate amounts of eosinophilic cytoplasm. (a) Sinusoidal infiltration pattern, (b) morphology (case 2, H&E, × 40 and × 500 magnification, respectively).

Cytologic features of ALK-DLBCL. (a) Case 3, (b) case 4 (H&E).
Immunohistochemistry
Table 3 provides a summary of the immunohistochemical results in our four cases. All tested cases were positive for CD138, monotypic cytoplasmic light chain and CD4 (Figure 3), CD45, EMA, VS38 and were negative for CD3, CD30, CD56, and TIA-1. Each case was positive for ALK in a granular cytoplasmic distribution (Figure 3). Three cases were CD20(−), while case 1 exhibited rare positive cells (Figure 4). Variable and scattered CD79a reactivity was seen in two cases. Two cases showed patchy CD57 positivity (Figure 4) and two cases exhibited rare CD43-positive cells. Occasional cytokeratin (AE1/AE3)(+) cells were seen in one case (Figure 4). OCT2, BOB.1, and MUM-1 were positive in case 4, and PAX-5 negative. CD38 was negative by immunohistochemistry in three cases, but positive by flow cytometry in one. IgA staining was equivocal in three cases. Cases 1, 2, 3, and 4 exhibited 50, 50, 80, and 50% proliferation indices by Ki-67 staining, respectively.

Typical immunohistochemical findings in ALK-DLBCL. Tumor cells are positive for (a, b) cytoplasmic, granular ALK, (c) CD138, (d) CD4 (cases 1, 4, 1, and 1, respectively).

Unusual immunohistochemical findings in occasional cases of ALK-DLBCL. (a) Rare CD20-positive tumor cells. (b) Scattered and variable CD57 membrane positivity. (c) Occasional cytokeratin-positive cells (cases 1, 2, and 2, respectively).
Flow Cytometry
Four-color flow cytometric analysis performed on tissue from case 2 revealed a 1% population of medium to large size cells (based on forward and side angle light scatter properties) expressing CD45, CD4, CD38 (moderate), and HLA-DR (Figure 5). The tumor cells were negative for CD2, CD3, CD5, CD7, CD8, CD10, CD14, CD19, CD20, CD23, FMC7, CD33, and surface kappa and lambda light chains.

Flow cytometric findings of ALK-DLBCL. A 1% population of medium to large cells (colored in red) expressing CD45, CD4, CD38 (moderate), and HLA-DR is identified (case 2). The tumor cells were negative for CD2, CD3, CD5, CD7, CD8, CD10, CD14, CD19, CD20, CD23, FMC7, CD33, and surface kappa and lambda light chains. Green—T cells; dark blue—monocytes; light blue—small, polytypic B cells; violet—plasma cells.
FISH for ALK Gene Rearrangement
FISH demonstrated an ALK gene rearrangement in case 4. Figure 6 demonstrates a clearly separated orange and green signal indicating a rearrangement of the ALK gene (arrows). The normal ALK gene signal is seen as overlapping/fusion of the orange and green signals (yellow). As the ALK probe is a break apart probe, the translocation partner remains unknown (eg Clathrin (CLTC) or NPM, etc.). However, given the cytoplasmic granular staining pattern with ALK immunohistochemistry, it is most likely that the partner is CLTC. FISH analysis on case 3 technically failed owing to lack of detectable hybridization signal.

Fluorescence in situ hybridization (FISH) of ALK-DLBCL showing ALK gene rearrangement using the ALK break apart probe from Vysis, Inc. The FISH pattern is positive for a rearrangement involving the ALK gene locus (arrows). The typical abnormal pattern would be expected to show 1R1G1F (one red and one green signal (abnormal) and one fused (yellow) (the remaining normal allele)). In this particular case, the majority of neoplastic cells also demonstrate an additional fused (yellow), signal indicating either a duplication of the ALK gene region or an additional copy of chromosome 2.
Discussion
We report four cases of anaplastic lymphoma kinase-positive diffuse large B-cell lymphoma (ALK-DLBCL) based on morphologic and immunophenotypic similarity to those previously described.1, 2, 3, 4, 5, 6 ALK-DLBCL was initially described by Delsol et al in 1997.1 Our study of four cases brings the total number of reported cases of ALK-DLBCL in the literature to 33.
ALK-DLBCL has a distinct morphologic appearance with immunoblastic/plasmablastic cytology with round, centrally to eccentrically located nuclei, prominent single central nucleoli, and moderate amounts of variably eosinophilic cytoplasm (Figures 1 and 2). A sinusoidal growth pattern may be seen (Figure 1a). Immunohistochemically, ALK-DLBCL shows features suggesting plasmacytic differentiation, with positivity for EMA, CD138, VS38c, and monotypic cytoplasmic light chain. The characteristic ALK staining is usually cytoplasmic and coarsely granular (attributed to the presence of the Clathrin-ALK fusion). Occasional cases with nuclear and cytoplasmic positivity have also been reported (Table 4). CD4, CD57, and CD45 are variably positive, and CD30, EBV, CD43, B-cell related antigens (CD20 and CD79a), and T-cell related antigens (CD3) are negative. Table 5 presents a summary of the frequency of common immunohistochemical findings. Delsol et al also found that a majority (5/7) of their cases expressed monotypic IgA lambda, which has also been subsequently reported by others. We and others have found that no cases express CD56 or TIA-1.4, 5, 6
Although a fairly typical immunohistochemical (plasmacytic) profile has been established for ALK-DLBCL, it is clear that some immunophenotypic heterogeneity exists. In one of our four cases, CD20 highlighted rare positive tumor cells, providing a helpful clue to the underlying B lineage. CD20 positivity, even focal, is distinctly unusual. We show one case with scattered cytokeratin (AE1/AE3)-positive tumor cells, which in conjunction with EMA positivity may lead to an erroneous interpretation of carcinoma. In addition, although usually negative, De Paepe et al3 report one of their three cases as being CD30 positive.
The overall morphologic and immunohistochemical features should allow for distinction of ALK-DLBCL from other entities including ALCL, plasmablastic lymphoma, plasmablastic myeloma, anaplastic variant of diffuse large B-cell lymphoma, and carcinoma. ALCL is typically CD30(+), of T-cell phenotype and would be negative for plasma cell markers (CD138) and immunoglobulin light chain. Plasmablastic lymphomas often occur in the oral region of human immunodeficiency virus-infected individuals, and are EBER positive and ALK negative.15 The anaplastic variant of DLBCL is usually strong CD20(+) and ALK(−). Plasmablastic myeloma has not been reported to be ALK positive, and would be associated with other features such as lytic bone lesions and a monoclonal protein in serum and/or urine.
The clinical features from the 33 reported cases of ALK-DLBCL are summarized in Tables 2 and 6. ALK-DLBCL spans all age groups with an overall male predominance (M:F ratio of 3:1). The M:F ratio is similar in children (7:3) and adults (18:5). Commonly reported clinical features included lymphadenopathy (27 cases), hepato- and/or splenomegaly (four cases), bony/CNS extension (four cases), mediastinal mass (four cases), and laryngeal/oral mass (three cases). Although only 33 cases of ALK-DLBCL have been reported thus far, higher stage disease at presentation (III–IV) appears to correlate with a poor response to multiagent lymphoma chemotherapy and an aggressive clinical course (Table 6). The overall median survival of high stage III/IV patients (N=13) was 11 months. Of the 11 patients reported as low stage I/II with at least 14 months follow-up, the average disease-free survival was 41 months (N=10). Only one was dead of disease after 14 months (Table 6).
The recent discovery of underlying ALK rearrangements in ALK-DLBCL is an important advance in our understanding of the pathogenesis of this lymphoma.2, 3, 4, 5, 6 The ALK gene located on chromosome 2p23 may be translocated to either the Clathrin (CLTC) gene locus on chromosome 17q23 or to the nucleophosmin (NPM) gene on chromosome 5q35, resulting in CLTC-ALK and NPM-ALK fusion products, respectively2, 3, 4, 5, 6, 8, 9, 10, 11 (Table 4). Both of these genetic rearrangements were originally identified in classic ALK(+) ALCL, with NPM-ALK being distinctly more common (70–80% of cases).16 As in ALCL, the ALK staining pattern in ALK-DLBCL appears to correlate with the type of underlying rearrangement. Cases with CLTC-ALK/t(2;17) rearrangement show a distinctly cytoplasmic and granular ALK staining pattern, whereas those cases with an NPM-ALK/t(2;5) rearrangement show both cytoplasmic and nuclear staining.2, 3, 4, 5, 6, 8, 9, 10, 11, 16 However, this correlation may be imperfect, as Onciu et al5 reported one case of ALK-DLBCL with NPM-ALK fusion that showed cytoplasmic ALK staining only. Thus, ALK gene rearrangements, originally thought to be uniquely associated with T-/null cell ALCL, have now been convincingly shown to occur in rare cases of B-cell lymphoma.2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13 Of note, prior to the initial series by Delsol et al,1 Arber et al17 in 1996 reported NPM/ALK fusion transcripts (by RT-PCR) in four of 33 cases of large B-cell lymphoma. Interestingly, and in contrast to the cases of ALK-DLBCL reported thus far, these four cases had a conventional B-cell immunophenotype (CD20+ and CD79a+). All cases were EMA(−) and one case had focal (<10%) CD30 positivity. ALK aberrations, specifically involving rearrangements of the CLTC gene, have also been identified in some cases of inflammatory myofibroblastic tumors (IMT).18 Thus, ALK overexpression likely contributes to the pathogenesis of a variety of otherwise unrelated neoplasms, ALK-DLBCL, ALCL, and IMT.
References
Delsol G, Lamant L, Mariame B, et al. A new subtype of large B-cell lymphoma expressing the ALK kinase and lacking the 2;5 translocation. Blood 1997;89:1483–1490.
Gascoyne R, Lamant L, Martin-Subero JI, et al. ALK-positive diffuse large B-cell lymphoma is associated with Clathrin-ALK rearrangements: report of six cases. Blood 2003;102:2568–2573.
De Paepe P, Baens M, van Krieken H, et al. ALK activation by the CLTC-ALK fusion is a recurrent event in large B-cell lymphoma. Blood 2003;102:2638–2641.
Chikatsu N, Kojima H, Suzukawa K, et al. ALK+, CD30−, CD20− large B-cell lymphoma containing anaplastic lymphoma kinase (ALK) fused to clathrin heavy chain gene (CLTC). Mod Pathol 2003;16:828–832.
Onciu M, Behm FG, Downing JR, et al. ALK-positive plasmablastic B-cell lymphoma with expression of the NPM-ALK fusion transcript: report of two cases. Blood 2003;102:2642–2644.
Adam P, Katzenberger T, Seeberger H, et al. A case of diffuse large B-cell lymphoma of plasmablastic type associated with the t(2;5)(p23;q35) chromosome translocation. Am J Surg Pathol 2003;27:1473–1476.
McManus DT, Catherwood MA, Carey PD, et al. ALK-positive diffuse large B-cell lymphoma of the stomach associated with a Clathrin-ALK rearrangement. Hum Pathol 2004;35:1285–1288.
Colomo L, Loong F, Rives S, et al. Diffuse large B-cell lymphomas with plasmablastic differentiation represent a heterogeneous group of disease entities. Am J Surg Pathol 2004;28:736–747.
Ishii K, Yamamoto Y, Nomura S . CD30-negative diffuse large B-cell lymphoma expressing ALK. Rinsho Ketsueki 2005;46:501–506.
Rudzski Z, Rucinska M, Jurczak W, et al. ALK-positive diffuse large B-cell lymphoma: two more cases and a brief literature review. Pol J Pathol 2005;56:37–45.
Gesk S, Gascoyne RD, Schnitzer B, et al. ALK-positive diffuse large B-cell lymphoma with ALK-Clathrin fusion belongs to the spectrum of pediatric lymphomas. Leukemia 2005;19:1839–1858.
Isimbaldi G, Bandiera L, d’Amore ES, et al. ALK-positive plasmablastic B-cell lymphoma with the clathrin-ALK gene rearrangement. Pediatr Blood Cancer 2006;46:390–391.
Bubala H, Maldyk J, Wlodarska I, et al. ALK-positive diffuse large B-cell lymphoma. Pediatr Blood Cancer 2006;46:649–653.
Reichard KK, McKenna RW, Kroft SH . Comparative analysis of light chain expression in germinal center cells and mantle cells of reactive lymphoid tissues: a four-color flow cytometric study. Am J Clin Pathol 2003;119:130–136.
Vega F, Chang CC, Medeiros LJ, et al. Plasmablastic lymphomas and plasmablastic plasma cell myelomas have nearly identical immunophenotypic profiles. Mod Pathol 2005;18:806–815.
Gatter KC, Warnke RA . Diffuse large B-cell lymphoma. In: Jaffe ES, Harris NL, Stein H, Vardiman JW (eds). World Health Organization Classification of Tumors: Tumors of the Haematopoietic and Lymphoid Tissues. IARC: Lyon, France, 2001, pp 174.
Arber DA, Sun LH, Weiss LM . Detection of the t(2;5) (p23;q35) chromosomal translocation in large B-cell lymphomas other than anaplastic large cell lymphoma. Hum Pathol 1996;27:590–594.
Bridge JA, Kanamori M, Ma Z, et al. Fusion of the ALK gene to the clathrin heavy chain gene, CLTC, in inflammatory myofibroblastic tumor. Am J Pathol 2001;159:411–415.
Acknowledgements
We thank Rodney Miller, MD, for kindly performing immunohistochemistry for VS38, CD38, and IgA; Randy Gascoyne, MD, for performing FISH analysis for the Clathrin/ALK rearrangement on case 3; and Rob McCauley, MD, for contributing case 4.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Reichard, K., McKenna, R. & Kroft, S. ALK-positive diffuse large B-cell lymphoma: report of four cases and review of the literature. Mod Pathol 20, 310–319 (2007). https://doi.org/10.1038/modpathol.3800742
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.3800742
Keywords
This article is cited by
-
ALK-positive large B-cell lymphoma: identification of EML4-ALK and a review of the literature focusing on the ALK immunohistochemical staining pattern
International Journal of Hematology (2016)
-
Mechanistic insight into ALK receptor tyrosine kinase in human cancer biology
Nature Reviews Cancer (2013)
-
ALK-positive large B-cell lymphomas express a terminal B-cell differentiation program and activated STAT3 but lack MYC rearrangements
Modern Pathology (2013)
