
Abstract
Significant intra- and interobserver variability exists in diagnosing and grading oral epithelial dysplasia. Mutations in the tumor-suppressor gene p16 are common in oral cavity dysplastic lesions, but whether immunohistochemical detection of the gene product p16INK4a (p16) can be used as a reliable biomarker for dysplasia is unclear. In total, 119 biopsy specimens representing various oral cavity sites and degrees of dysplasia were retrieved from the pathology files of Emory University Hospital. Formalin-fixed, paraffin-embedded sections were stained with hematoxylin and eosin (H&E) and with a monoclonal antibody to p16 (LabVision Corporation, Clone JC2). A blinded review of the H&E slides and the pattern and degree of p16 expression was independently performed by two pathologists. A consensus was obtained when diagnoses differed. Morphologic diagnoses were then compared to p16 immunohistochemical expression. Overall, 61/119 (51%) cases showed no p16 immunoreactivity, including 12/33 (36%) cases of no dysplasia, 11/28 (39%) cases of mild dysplasia, and 38/58 (66%) cases of moderate/severe dysplasia. The remaining cases showed p16 expression limited to the basal and suprabasal nuclei and generally confined to the lower one-third of the epithelium. A logistic regression model showed a trend toward absent p16 expression with increasing severity of dysplasia (P=0.006). Decreased expression of p16 in dysplastic lesions, as found in this study, may reflect the biologic events involving loss of p16 gene function in the pathogenesis of oral cancer. Our findings suggest that p16 immunohistochemistry is not helpful in differentiating dysplastic from nondysplastic mucosa in oral cavity biopsies, and thus is not a reliable biomarker for use in routine clinical practice.
Similar content being viewed by others

Main
Worldwide there were an estimated 274 000 new cases of oral cavity cancer in 2002, with almost two-thirds occurring in men,1 and the overwhelming majority are squamous cell carcinomas. For many years it has been known that invasive squamous cell carcinoma is preceded by a progressive accumulation of genetic mutations within the epithelial cells.2 Premalignant changes are often clinically apparent as leukoplakia or erythroplakia, and histologically are identified as dysplasia. In a review by Barnes, the risk for developing invasive squamous cell carcinoma in laryngeal mucosa for patients with mild, moderate, and severe dysplasia was 5.7, 22.5, and 28.4%, respectively.3 Thus, there is a five-fold increased risk of developing invasive carcinoma for severe compared to mild dysplasia, which highlights the need for accurate histologic grading of clinically suspicious lesions.
Although multiple different grading systems have been proposed, the generally preferred scheme for dysplastic lesions of the oral cavity includes mild, moderate, and severe dysplasia. Grading of oral dysplasia is based on architectural, cytomorphologic, and maturation abnormalities including irregular epithelial stratification, drop-shaped rete processes, loss of polarity of the basal cells, single cell keratinization, increased nuclear/cytoplasmic ratio, increased number of mitotic figures, cellular and nuclear pleomorphism, enlarged nucleoli, and loss of cellular cohesion.4 While these histologic features are widely known, multiple studies have shown that intra- and interobserver agreement in the diagnosis and grading of oral dysplasia is generally poor.5, 6, 7, 8, 9
Tumor-suppressor gene inactivation has been associated with oral dysplasia and squamous cell carcinoma, most commonly involving loci on chromosomes 3p, 9p, and 17p, although many others have been described.2, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23 The function of the tumor-suppressor genes located on chromosome 3p are currently poorly defined, whereas the functions of p16 (also known as CDKN2A, located on 9p21) and p53 (located on 17p13) are well established. Inactivation of these genes causes altered expression of their respective gene products p16INK4a (p16) and p53, which suggests that they may be useful biomarkers for oral cavity dysplasia and carcinoma.20, 21, 24, 25, 26, 27
Immunohistochemical detection of p16 has been shown to be a reliable marker for squamous dysplasia in the uterine cervix, where the protein is frequently overexpressed as a result of infection with oncogenic strains of human papillomavirus (HPV).28, 29, 30, 31, 32 More recently, a study showed immunohistochemical overexpression of p16 in dysplastic lesions of the oral cavity, which suggests that it may be a useful ancillary test for diagnosing dysplasia in this location.33 However, several other studies have shown decreased p16 expression in oral premalignant and malignant lesions by immunohistochemistry, which has been attributed to gene inactivating mechanisms such as homozygous deletion, point mutation, and promoter hypermethylation.20, 21, 27, 34 The purpose of the current study was to further investigate p16 immunohistochemistry as a potential biomarker for dysplasia using a large series of oral cavity biopsies with and without dysplasia with the goal of determining its usefulness for routine clinical practice.
Materials and methods
Case Selection
This study was approved by the Emory University Institutional Review Board. In total, 119 biopsy specimens accessioned between April 2002 and July 2005 representing various oral cavity sites and degrees of dysplasia (Table 1) were retrieved from the pathology files of Emory University Hospital. Only well-characterized cases of normal and dysplastic mucosa were selected so that the patterns of p16 staining could be reliably compared to degree of dysplasia. Poorly oriented biopsies and biopsies exhibiting ulceration were excluded.
p16 Immunohistochemistry
For each case, a 4-μm-thick section of formalin-fixed, paraffin-embedded tissue was stained with hematoxylin and eosin (H&E), and an adjacent section was immunostained for the presence of p16 (mouse monoclonal antibody, clone 16P04, 1:40 dilution, LabVision, Fremont, CA, USA) using heat-induced antigen retrieval, horseradish peroxidase (HRP)-labeled polymer conjugated with secondary antibodies (Dako Envision System, Carpinteria, CA, USA), and the Dako Autostainer (Dako, Carpinteria, CA, USA). A section of vaginal squamous cell carcinoma was used as the positive control. Negative controls had primary antibody replaced by buffer.
Tissue sections were deparaffinized and rehydrated. Antigen retrieval was performed in citrate buffer (pH 6) using an electric pressure cooker for 5 min at 120°C, with cooling for 10 min before immunostaining. Tissues were then exposed to 3% hydrogen peroxide for 5 min to block endogenous peroxidase, followed by appropriately characterized and diluted primary antibody for 30 min, HRP-labeled polymer for 30 min, diaminobenzidine as chromogen for 5 min, and Dako automation hematoxylin as counterstain for 15 min. These incubations were performed at room temperature. Between incubations, sections were washed with Tris-buffered saline (TBS) buffer. Coverslipping was performed using the Tissue Tek SCA automatic coverslipper (Sakura Finetek USA Inc., Torrance, CA, USA).
Evaluation of Cases for Dysplasia and Immunohistochemical Staining
Each recut H&E slide was independently examined by two pathologists (SDB and SL) and graded as no, mild, moderate, or severe dysplasia without knowledge of the original sign-out diagnosis. The percentage of cases showing exact agreement between the two observers was calculated, as were levels of disagreement. A consensus grade was determined by joint review for cases in which individual diagnoses differed. However, consensus was not attempted when one pathologist diagnosed moderate dysplasia and the other diagnosed severe dysplasia because there is relatively less clinical importance in distinguishing these two grades since they are often managed similarly.
p16-stained slides were reviewed independently by the same two pathologists without knowledge of the H&E grade. Positive cases were defined as having five or more squamous epithelial cells with staining of the nucleus, cytoplasm, or both. Positive cases were divided into two semiquantitative groups: basal layer staining, in which staining was confined to the basal cells with no staining or very rare staining of more superficial cells; and basal and suprabasal layer staining, defined as positivity of cells in both the basal and suprabasal layers. Consensus was obtained when individual interpretations differed.
To verify the technical validity of our results, p16 immunohistochemical staining was repeated for 15 of the 119 cases using the identical technique as described above. Cases were randomly chosen from those diagnosed as moderate/severe dysplasia. Staining patterns for these cases were interpreted by one of us (SL) without knowledge of the previous staining pattern. The same criteria were used for interpretation as described above.
Statistical Analysis
For statistical analyses, p16-negative cases were compared to p16-positive cases for each degree of dysplasia. Positive cases were grouped together because the difference between basal layer staining and basal plus suprabasal layer staining, while potentially informative histologically, is undefined in terms of p16 inactivation. A χ2 test of independence was used to test the null hypothesis that immunohistochemical expression of p16 is unrelated to degree of dysplasia. Trends in the data were assessed using a proportional odds ordinal logistic regression model. A P-value of <0.05 was used to indicate statistical significance.
Results
Dysplasia Grading
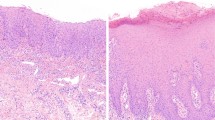
Consensus histologic grading for the 119 cases included in this study showed there to be 33 cases with no dysplasia, 28 cases with mild dysplasia, and 58 cases with moderate/severe dysplasia (Table 2, Figure 1). There was exact agreement in grade of dysplasia between the two observers for 69 of 119 cases (58%) including 28 cases diagnosed as no dysplasia, 12 cases of mild dysplasia, nine cases of moderate dysplasia, and 20 cases of severe dysplasia. There was agreement within one histologic grade for 117 of 119 cases (98%). Overall, this level of agreement is higher than that typically reported in the literature, which likely reflects the fact that only cases of well-characterized dysplasia were included during case selection.

Representative H&E consensus histologic grades of oral cavity biopsies: (a) no dysplasia; (b) mild dysplasia; (c) moderate/severe dysplasia.
p16 Immunohistochemistry
Overall, 61 of 119 cases (51%) were negative for p16 immunoreactivity, including the majority of cases diagnosed as moderate/severe dysplasia (Table 2). In the remaining cases, p16 expression was limited to the basal and suprabasal cell layers and generally was confined to the lower one-third of the epithelium. Representative examples of the immunohistochemical staining patterns are shown in Figure 2. The positive control was strongly and diffusely positive (Figure 2). χ2 analysis showed p16 expression to be dependent on severity of dysplasia (χ2=9.26, df=2, P=<0.01) with a significant trend toward absent p16 expression with increasing severity of dysplasia (logistic regression model, β=−0.62, P=0.006). For the 15 cases in which p16 immunohistochemistry was repeated, each showed a staining pattern that was similar to the original run.

Representative immunohistochemical staining patterns of oral cavity biopsies using a p16 monoclonal antibody: (a) basal layer staining; (b) basal and suprabasal layer staining. (c) The most strongly positive case showed focal areas with p16 staining of the mid to upper cell layers; however, adjacent areas on the same slide showed no p16 expression. (d) The p16 positive control is strongly and diffusely positive (vaginal squamous cell carcinoma).
Discussion
Oncogenesis in the oral cavity is widely believed to result from cumulative genetic alterations that cause a step-wise transformation of the mucosa from normal to dysplastic to invasive carcinoma.2 Attempts have been made to correlate the specific genetic changes with histopathological progression, but this has remained a challenge since numerous genes are involved.2 In a review, Patel et al17 summarized the chromosomal abnormalities that are commonly found in oral cancer, including losses of 3p, 4q, 5q21–22, 8p21–23, 9p21–22, 11q13, 11q23, 13q, 14q, 17p, 18q, and 22q. Of these, loss of chromosomal 9p is frequently reported to be the most common genetic abnormality in oral dysplasia and carcinoma.2, 11, 15 Various studies have shown loss of heterozygosity on 9p21 in 20% of benign squamous hyperplasias,2 28–71% of dysplastic lesions,2, 11, 14, 22 and 19–73% of invasive squamous cell carcinomas.2, 11, 13, 14, 15, 25, 35 In addition, because invasive carcinomas often do not exhibit increased loss of heterozygosity compared to preinvasive lesions, loss of 9p is considered an early event in tumor progression.2, 14
The tumor-suppressor gene p16 is localized on 9p21, and its inactivation is considered to be a significant event in development of many tumor types including oral carcinoma,36 although one study showed that additional tumor-suppressor genes may exist at this locus.35 The protein gene product p16 normally binds to cyclin-dependent kinases (cdk) 4 and 6, inhibiting their association with cyclin D1. The inhibition of the cdk 4/6-cyclin D1 complex prevents phosphorylation of the retinoblastoma protein (pRb) leading to inhibition of cell cycle progression through G1- to S-phase.10 While inactivation of pRb is variable in oral squamous cell carcinoma, inactivation of p16 is common and usually occurs via homozygous deletion, point mutation, or promoter hypermethylation.13, 16, 20, 21, 27, 34, 37, 38
The frequent occurrence of p16 inactivation during early carcinogenesis has led to its investigation as a surrogate marker for dysplasia. The diagnosis of oral dysplasia is currently dependent on interpretation of H&E-stained slides, but this is known to be subjective with considerable intra- and interobserver variability.5, 6, 7, 8, 9 Immunohistochemical evaluation of oral premalignant and malignant lesions for p16 expression using an anti-p16 antibody has given variable results with some studies showing decreased expression20, 21, 27, 34 and others showing overexpression.33, 39 The two studies showing overexpression and one study showing decreased expression27 each utilized a polyclonal antibody (C-20, Santa Cruz Biotechnology, Santa Cruz, CA, USA), while the remaining studies used various monoclonal antibodies. In our experience, the C-20 polyclonal antibody produces nonspecific staining including prominent muscle staining and positivity of both dysplastic and nondysplastic oral mucosa (unpublished data). Prior studies have shown excellent concordance between p16 gene inactivation as determined by molecular methods and p16 protein expression detected using immunohistochemistry.20, 21, 27, 34 Thus, the frequent loss of the p16 gene in dysplastic mucosa would be expected to result in a decrease in p16 expression compared to normal mucosa.
In the current study, we used a monoclonal antibody to investigate the immunohistochemical expression of p16 in oral mucosa biopsies showing varying degrees of dysplasia. Our data show a significant trend toward absent expression of p16 with increasing severity of dysplasia. However, loss of p16 expression was not limited to dysplastic lesions, since a significant percentage of nondysplastic mucosa was also negative. Furthermore, when present, staining was confined to the basal and suprabasal cell layers in both normal and markedly dysplastic mucosa with no cases showing full-thickness positivity. These findings indicate heterogeneous expression of p16 within morphologically homogeneous tissue. In addition, our data shows that p16 expression cannot reliably differentiate normal from dysplastic mucosa.
Our finding of variable p16 expression in oral cavity biopsies is in contrast to the uterine cervix where squamous dysplastic lesions frequently overexpress p16 as a result of infection with high-risk HPV types. This has resulted in the use of p16 immunohistochemistry as a biomarker of cervical dysplasia.28, 29, 30, 31, 32 In the cervix, p16 overexpression is thought to be the result of inactivation of pRb by the HPV E7 oncoprotein. The interaction of E7 with pRb results in release of the transcription factor E2F from the active pRb-E2F complex. As pRb-E2F normally inhibits transcription of the p16 gene, expression of HPV E7 results in excessive and deregulated transcription and translation of p16.40 Thus, the mechanism for p16 overexpression in HPV-related cervical dysplastic lesions is thought to be different from the mechanism of p16 inactivation, which is more common in oral cavity dysplasia.
Using monoclonal p16 immunohistochemistry in oral malignancies, Fregonesi et al41 showed a strong association between overexpression of p16 and infection with high-risk HPV types. Their study demonstrated diffuse p16 positivity in 69% of HPV 16/18 cases compared to 4% of HPV-negative cases and 0% of HPV 6/11 cases, although focal or sporadic staining was seen in many of the HPV-negative and HPV 6/11 cases.41 In addition, a study by Saito et al26 showed overexpression of p16 in 45% of oral verrucous carcinomas, which are etiologically often associated with HPV infection, compared to only 11% of cases of usual squamous cell carcinoma. These findings have led to the suggestion that overexpression of p16 may serve as a biomarker for HPV-induced oral dysplasia or carcinoma. However, the incidence of HPV infection in oral mucosa is variable,42, 43, 44, 45 which implies that immunohistochemistry is likely to be unreliable without knowledge of HPV status. As HPV status was not evaluated in the current study, it is unclear whether our results are associated with a low prevalence of high-risk HPV infection in the population studied.
Decreased immunohistochemical expression of p16 in dysplastic lesions of the oral cavity, as found in this study, may be due to p16 inactivation, although this was not tested. Additional studies investigating p16 gene inactivation and high-risk HPV status concurrently with immunohistochemical detection of p16 would be helpful in clarifying the patterns of p16 expression observed in dysplastic mucosae. Regardless of the mechanism involved, our findings suggest that p16 immunohistochemistry is not helpful in differentiating dysplastic from nondysplastic mucosa in oral cavity biopsies, and thus is not a reliable biomarker for use in routine clinical practice.
References
Parkin DM, Bray F, Ferlay J, et al. Global cancer statistics, 2002. CA Cancer J Clin 2005;55:74–108.
Califano J, van der Riet P, Westra W, et al. Genetic progression model for head and neck cancer: implications for field cancerization. Cancer Res 1996;56:2488–2492.
Barnes L . Diseases of the larynx, hypopharynx and esophagus. In: Barnes L (ed). Surgical Pathology of the Head and Neck, 2nd edn. Marcel Dekker Inc.: New York, 2001, pp 127–237.
Kramer IR, Lucas RB, Pindborg JJ, et al. Definition of leukoplakia and related lesions: an aid to studies on oral precancer. Oral Surg Oral Med Oral Pathol 1978;46:518–539.
Brothwell DJ, Lewis DW, Bradley G, et al. Observer agreement in the grading of oral epithelial dysplasia. Commun Dent Oral Epidemiol 2003;31:300–305.
Sudbø J, Bryne M, Johannessen AC, et al. Comparison of histological grading and large-scale genomic status (DNA ploidy) as prognostic tools in oral dysplasia. J Pathol 2001;194:303–310.
Abbey LM, Kaugars GE, Gunsolley JC, et al. Intraexaminer and interexaminer reliability in the diagnosis of oral epithelial dysplasia. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 1995;80:188–191.
Karabulut A, Reibel J, Therkildsen MH, et al. Observer variability in the histologic assessment of oral premalignant lesions. J Oral Pathol Med 1995;24:198–200.
Pindborg JJ, Reibel J, Holmstrup P . Subjectivity in evaluating oral epithelial dysplasia, carcinoma in situ and initial carcinoma. J Oral Pathol 1985;14:698–708.
Nagpal JK, Das BR . Oral cancer: reviewing the present understanding of its molecular mechanism and exploring the future directions for its effective management. Oral Oncol 2003;39:213–221.
El-Naggar AK, Hurr K, Batsakis JG, et al. Sequential loss of heterozygosity at microsatellite motifs in preinvasive and invasive head and neck squamous carcinoma. Cancer Res 1995;55:2656–2659.
Williams HK . Molecular pathogenesis of oral squamous carcinoma. Mol Pathol 2000;53:165–172.
Sartor M, Steingrimsdottir H, Elamin F, et al. Role of p16/MTS1, cyclin D1 and RB in primary oral cancer and oral cancer cell lines. Br J Cancer 1999;80:79–86.
van der Riet P, Nawroz H, Hruban RH, et al. Frequent loss of chromosome 9p21-22 early in head and neck cancer progression. Cancer Res 1994;54:1156–1158.
Nawroz H, van der Riet P, Hruban RH, et al. Allelotype of head and neck squamous cell carcinoma. Cancer Res 1994;54:1152–1155.
Prime SS, Eveson JW, Guest PG, et al. Early genetic and functional events in the pathogenesis of oral cancer. Radiat Oncol Investig 1997;5:93–96.
Patel V, Leethanakul C, Gutkind JS . New approaches to the understanding of the molecular basis of oral cancer. Crit Rev Oral Biol Med 2001;12:55–63.
Partridge M, Emilion G, Peteromichelakis S, et al. Location of candidate tumour suppressor gene loci at chromosomes 3p, 8p and 9p for oral squamous cell carcinomas. Int J Cancer 1999;83:318–325.
Field JK . Oncogenes and tumour-suppressor genes in squamous cell carcinoma of the head and neck. Eur J Cancer B Oral Oncol 1992;28B:67–76.
Reed AL, Califano J, Cairns P, et al. High frequency of p16 (CDKN2/MTS-1/INK4A) inactivation in head and neck squamous cell carcinoma. Cancer Res 1996;56:3630–3633.
Papadimitrakopoulou V, Izzo J, Lippman SM . Frequent inactivation of p16INK4a in oral premalignant lesions. Oncogene 1997;14:1799–1803.
Mao L, Lee JS, Fan YH, et al. Frequent microsatellite alterations at chromosomes 9p21 and 3p14 in oral premalignant lesions and their value in cancer risk assessment. Nat Med 1996;2:682–685.
Zhang L, Rosin MP . Loss of heterozygosity: a potential tool in management of oral premalignant lesions? J Oral Pathol Med 2001;30:513–520.
Lee NK, Ye Y, Chen J, et al. p53, retinoblastoma, and human papillomavirus in squamous cell carcinoma and adjacent normal mucosa of the upper aerodigestive tract. Arch Otolaryngol Head Neck Surg 1993;119:1125–1131.
Olshan AF, Weissler MC, Pei H, et al. Alterations of the p16 gene in head and neck cancer: frequency and association with p53, PRAD-1 and HPV. Oncogene 1997;14:811–818.
Saito T, Nakajima T, Mogi K . Immunohistochemical analysis of cell cycle-associated proteins p16, pRb, p53, p27 and Ki-67 in oral cancer and precancer with special reference to verrucous carcinomas. J Oral Pathol Med 1999;28:226–232.
Pande P, Mathur M, Shukla NK, et al. pRb and p16 protein alterations in human oral tumorigenesis. Oral Oncol 1998;34:396–403.
Klaes R, Friedrich T, Spitovsky D, et al. Overexpression of p16INK4a as a specific marker for dysplastic and neoplastic epithelial cells of the cervix uteri. Int J Cancer 2001;92:276–284.
Klaes R, Benner A, Friedrich T, et al. p16INK4a immunohistochemistry improves interobserver agreement in the diagnosis of cervical intraepithelial neoplasia. Am J Surg Pathol 2002;26:1389–1399.
Sano T, Masuda N, Oyama T, et al. Overexpression of p16 and p14ARF is associated with human papillomavirus infection in cervical squamous cell carcinoma and dysplasia. Pathol Int 2002;52:375–383.
Dray M, Russell P, Dalrymple C, et al. p16INK4a as a complementary marker of high-grade intraepithelial lesions of the uterine cervix. I: Experience with squamous lesions in 189 consecutive cervical biopsies. Pathology 2005;37:112–124.
Murphy N, Ring M, Heffron CC, et al. p16INK4a, CDC6, and MCM5: predictive biomarkers in cervical preinvasive neoplasia and cervical cancer. J Clin Pathol 2005;58:525–534.
Gologan O, Barnes EL, Hunt JL . Potential diagnostic use of p16INK4a, a new marker that correlates with dysplasia in oral squamoproliferative lesions. Am J Surg Pathol 2005;29:792–796.
Ai L, Stephenson KK, Ling W, et al. The p16 (CDKN2a/INK4a) tumor-suppressor gene in head and neck squamous cell carcinoma: a promoter methylation and protein expression study in 100 cases. Mod Pathol 2003;16:944–950.
Waber P, Dlugosz S, Cheng Q, et al. Genetic alterations of chromosome band 9p21-22 in head and neck cancer are not restricted to p16INK4a. Oncogene 1997;15:1699–1704.
Cairns P, Polascik TJ, Eby Y, et al. Frequency of homozygous deletion at p16/CDKN2 in primary human tumours. Nat Genet 1995;11:210–212 [letter].
Pavelic ZP, Lasmar M, Pavelic L, et al. Absence of retinoblastoma gene product in human primary oral cavity carcinomas. Eur J Cancer B Oral Oncol 1996;32B:347–351.
Yoo GH, Xu HJ, Brennan JA, et al. Infrequent inactivation of the retinoblastoma gene despite frequent loss of chromosome 13q in head and neck squamous cell carcinoma. Cancer Res 1994;54:4603–4606.
Paradiso A, Ranieri G, Stea B, et al. Altered p16INK4a and Fhit expression in carcinogenesis and progression of human oral cancer. Int J Oncol 2004;24:249–255.
Vinokurova S, Wentzensen N, von Knebel Doeberitz M . Analysis of p16INK4a and integrated HPV genomes as progression markers. In: Davy C, Doorbar J (eds). Methods in Molecular Medicine, Vol 119. Human Papillomaviruses: Methods and Protocols. Humana Press Inc.: Totowa, NJ, 2005, pp 73–83.
Fregonesi PA, Teresa DB, Duarte RA, et al. p16INK4a immunohistochemical overexpression in premalignant and malignant oral lesions infected with human papillomavirus. J Histochem Cytochem 2003;51:1291–1297.
González-Moles MA, Ruiz-Avila I, González-Moles S, et al. Detection of HPV DNA by in situ hybridization in benign, premalignant and malignant lesions of the oral mucosa. Bull Group Int Rech Sci Stomatol Odontol 1994;37:79–85.
Bouda M, Gorgoulis VG, Kastrinakis NG, et al. ‘High risk’ HPV types are frequently detected in potentially malignant and malignant oral lesions, but not in normal oral mucosa. Mod Pathol 2000;13:644–653.
Syrjänen S . Human papillomavirus (HPV) in head and neck cancer. J Clin Virol 2004;32(Suppl 1):S59–S66.
Sugiyama M, Bhawal UK, Dohmen T, et al. Detection of human papillomavirus-16 and HPV-18 DNA in normal, dysplastic, and malignant oral epithelium. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 2003;95:594–600.
Acknowledgements
We thank Melanie Pearson, PhD and Brian Schmotzer, MS for their assistance with statistical analyses, and Susan Muller, DMD for her critical review of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
This work was presented as a proffered paper at the 95th annual meeting of the United States and Canadian Academy of Pathology, Atlanta, GA, 11–17 February 2006 (Mod Pathol 2006;19 (Supp 1): 204A).
Duality of interest
None declared.
Rights and permissions
About this article
Cite this article
Bradley, K., Budnick, S. & Logani, S. Immunohistochemical detection of p16INK4a in dysplastic lesions of the oral cavity. Mod Pathol 19, 1310–1316 (2006). https://doi.org/10.1038/modpathol.3800649
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.3800649
Keywords
This article is cited by
-
Lymphoepithelial carcinoma of the parotid gland: a unique example showing p16 immunoreactivity
Medical Molecular Morphology (2021)
-
Molecular Analysis as a Guide to Determining the Extent and Pathophysiology of Perilesional Tissues in Oral Epithelial Dysplasias
Journal of Maxillofacial and Oral Surgery (2020)
-
Morphometric analysis of Ki-67 and p16 expression in laryngeal precursor lesions
European Archives of Oto-Rhino-Laryngology (2013)
-
Scoring mechanisms of p16INK4a immunohistochemistry based on either independent nucleic stain or mixed cytoplasmic with nucleic expression can significantly signal to distinguish between endocervical and endometrial adenocarcinomas in a tissue microarray study
Journal of Translational Medicine (2009)
