
Abstract
The human epidermal growth factor receptor (HER) family of receptor tyrosine kinase has been extensively studied in breast cancer; however, systematic studies of EGFR gene amplification and protein overexpression in breast carcinoma are lacking. We studied EGFR gene amplification by chromogenic in situ hybridization (CISH) and protein expression by immunohistochemistry in 175 breast carcinomas, using tissue microarrays. Tumors with >5 EGFR gene copies per nucleus were interpreted as positive for gene amplification. Protein overexpression was scored according to standardized criteria originally developed for HER-2. EGFR mRNA levels, as measured by Affymetrix U133 Gene Chip microarray hybridization, were available in 63 of these tumors. HER-2 gene amplification by fluorescence in situ hybridization (FISH) and protein overexpression by immunohistochemistry were also studied. EGFR gene amplification (copy number range: 7–18; median: 12) was detected in 11/175 (6%) tumors, and protein overexpression was found in 13/175 (7%) tumors. Of the 11 tumors, 10 (91%) with gene amplification also showed EGFR protein overexpression (2+ or 3+ by immunohistochemistry). The EGFR mRNA level, based on Affymetrix U133 chip hybridization data, was increased relative to other breast cancer samples in three of the five tumors showing gene amplification. Exons 19 and 21 of EGFR, the sites of hotspot mutations in lung adenocarcinomas, were screened in the 11 EGFR-amplified tumors but no mutations were found. Three of these 11 tumors also showed HER-2 overexpression and gene amplification. Approximately 6% of breast carcinomas show EGFR amplification with EGFR protein overexpression and may be candidates for trials of EGFR-targeted antibodies or small inhibitory molecules.
Similar content being viewed by others


Main
The epidermal growth factor receptor (EGFR, HER-1, c-erbB-1) is one of the four transmembrane growth factor receptor proteins that share similarities in structure and function. Together, this group comprises the human epidermal growth factor receptor (HER) (c-erbB) family of receptor tyrosine kinases. The EGFR gene is located on the short arm of chromosome 7 and encodes a 170 kDa transmembrane protein consisting of an extracellular EGF-binding domain, a short transmembrane region, and an intracellular domain with ligand-activated tyrosine kinase activity.1 Two ligands can activate EGFR: epidermal growth factor (EGF) and transforming growth factor-alpha (TGF-α). Ligand binding to EGFR results in receptor homo- or hetero-dimerization (with one of the HER family of receptor tyrosine kinases) followed by autophosphorylation of the tyrosine kinase domain.2 Phosphorylated tyrosine residues serve as binding sites for the recruitment of signal transducers and activators of intracellular substrates. The Ras–Raf mitogen-activated protein kinase pathway and the phosphatidyl inositol 3′ kinase and Akt pathway are the major signaling routes for the HER family, including EGFR.3, 4, 5, 6 These pathways control several important biologic processes, including cellular proliferation, angiogenesis and inhibition of apoptosis.7
The interest in EGFR is further enhanced by the availability and FDA approval of specific EGFR tyrosine kinase inhibitors (eg, gefitinib). Many of these studies have focused on lung cancer, where approximately 10% of patients have a rapid and often dramatic clinical response.8, 9, 10 These gefitinib-responsive lung cancers have been found to contain somatic mutations in the tyrosine kinase domain of the EGFR gene.8, 9, 10 The data regarding the presence or absence of EGFR gene amplification in other tumor types, and their response to these EGFR tyrosine kinase inhibitors are still limited. EGFR protein overexpression has been reported to occur in 16–36% of breast cancers; however, systematic studies evaluating gene amplification, mRNA expression and protein expression in the same set of cases are lacking.11, 12, 13 In order to address this issue, we studied 175 breast cancers for the presence of EGFR gene amplification. In addition, we analyzed EGFR protein expression, HER-2 protein expression and gene amplification in these tumors. We also examined EGFR transcript levels in a subset of these tumors by Affymetrix U133 chip hybridization and performed a mutational screen of the EGFR-amplified cases.
Materials and methods
Case Selection and Tissue Microarray Construction
In all, 188 randomly selected invasive breast carcinomas were included in this study. Tissue microarrays were created using 0.6 mm tissue cores as previously described.14, 15, 16, 17, 18 An H&E-stained section was evaluated for the presence of invasive breast carcinoma and the area to be used for creation of the tissue microarrays was marked on the slide and the donor block. Three to four cores from different areas of the tumor were sampled for each tumor.
Histologic Examination
Histologic assessment of tumor type and grade were routinely performed on 4–5 μm thick H&E sections of formalin-fixed paraffin-embedded tumors. The nuclear grades of invasive ductal and lobular carcinomas were designated as follows: grade 1, small, regular uniform cells; grade 2, moderate increase in size and variability; grade 3, marked variation in size and shape. The architectural grades of invasive ductal carcinomas were designated as follows: grade 1, well developed (>75%) tubule formation; grade 2, moderate (10–75%) tubule formation; grade 3, little or no (<10%) tubule formation.
Immunohistochemistry
Tissue microarray sections (4–5 μm thick) were used for all immunohistochemical analyses. The Ventana CONFIRM™ antiestrogen receptor (clone 6F11) and antiprogesterone receptor (clone 16) monoclonal antibodies were used for immunohistochemical analyses of estrogen receptor and progesterone receptor, respectively, performed on the Ventana automated slide stainers according to the manufacturer's instructions (Ventana Inc., Tucson, AZ, USA). The estrogen receptor or progesterone receptor results were manually screened and were interpreted as positive when more than 10% of tumor cells showed positive nuclear staining. HER-2 immunohistochemistry was performed using the HercepTest™ kit (DAKO Corp, Carpinteria, CA, USA) and EGFR immunohistochemistry was performed using a monoclonal EGFR antibody (Clone 31G7, Zymed Laboratories Inc., South San Francisco, CA, USA) according to the manufacturer's instructions; both HER-2 and EGFR results were interpreted manually as follows: 0, no membrane staining; 1+, faint, partial membrane staining; 2+, weak, complete membrane staining in >10% of invasive cancer cells; 3+, intense complete membrane staining in >10% of invasive cancer cells. The highest immunohistochemical score obtained among different cores of the same tumor was used as the final immunohistochemical result of that tumor.
Chromogenic In Situ Hybridization
Chromogenic in situ hybridization (CISH) for EGFR gene was performed according to the manufacturer's instructions. Briefly, the tissue microarray sections were incubated at 55°C overnight. The slides were deparaffinized in xylene and graded ethanols. Heat pretreatment was carried out in the pretreatment buffer (Zymed Laboratories Inc.) at 98–100°C for 15 min. The tissue was digested with pepsin for 10 min at room temperature. After application of Zymed SpotLight® digoxigenin labeled EGFR probe (Zymed Laboratories Inc.), the slides were cover-slipped and edges sealed with rubber cement. The slides were heated at 95°C for 5 min followed by overnight incubation at 37°C using a moisturized chamber. Posthybridization wash was performed the next day and followed by immunodetection using the CISH™ polymer detection kit (Zymed Laboratories Inc.). The CISH signals were counted in at least 30 nuclei with a light microscope using a × 40 objective. A tumor was interpreted as positive for gene amplification when the average number of gene copies was >5 per nucleus.
Fluorescence In Situ Hybridization
Fluorescence in situ hybridization (FISH) for HER-2 was performed using the PathVysion HER-2 probe kit (Vysis Inc. Downers Grove, IL, USA) as previously described.17 The signal enumeration was performed under × 1000 magnification. The number of chromosome 17 signals, HER-2 signals, and number of tumor nuclei scored were recorded for each core. At least 30 cells were counted per tissue core. Tumors were interpreted as amplified when the ratio of HER-2/chromosome17 signals was ≥2.0. The average ratio of different cores from the same tumor was used as the final score for determination of gene amplification status of that particular tumor.
EGFR mRNA Expression
EGFR mRNA levels were determined in a subset of cases using Affymetrix human genome U133 GeneChip® expression arrays. RNA extraction, RNA target synthesis, and target labeling were performed as previously described.19 Gene expression analysis was carried out using the Affymetrix U133A human gene array, which has 22 283 features for individual gene/EST clusters, using instruments and protocols recommended by the manufacturer. For each gene on every sample we extracted two response measures, the Average Difference and Absolute Call, as determined by the default settings of Affymetrix Microarray Suite 5.0. Expression values on each array were multiplicatively scaled to have an average expression of 500 across the central 96% of all genes on the array. Calculations of relative EGFR transcript levels were based on data from Affymetrix probe set 201984_s_at.
EGFR Mutation Analysis
Selected cases were analyzed for the presence of hotspot mutations in exon 19 (short in-frame deletions) and exon 21 (L858R mutation) that together account for approximately 90% of EGFR mutations detected in lung cancers.8, 9, 10 Exon 19 deletions were studied by length analysis of fluorescently labeled polymerase chain reaction (PCR) products on a capillary electrophoresis device, and the exon 21 L585R mutation was detected by PCR followed by Sau96I restriction enzyme digestion, based on a new Sau96I site created by the L585R mutation (2819T>G), followed by capillary electrophoresis of the Sau96I-digested fluorescently labeled PCR products. These sensitive assays can detect mutations in the presence of up to 90% non-neoplastic cells and are described in detail elsewhere.20
Results
We obtained both CISH and immunohistochemistry EGFR data on 175 of the 188 breast cancers. Nine tumors failed both CISH and immunohistochemistry, four additional tumors failed immunohistochemistry alone. The reasons for failure were a complete loss of tissue cores from the tissue microarrays, less than 30 tumor cells available for scoring, and absence of hybridization signals. The absence of signals probably resulted from under- or over-digestion since tissue digestion for a particular tumor cannot be adjusted on a tissue microarray.
EGFR gene copy number ranged from 2 to 18 in the samples studied. Copy number greater than 5 was considered amplified and identified in 11/175 (6%) tumors (Table 1). The gene copy number in amplified tumors ranged from 7 to 18 (mean: 12.1; median: 12) and in nonamplified tumors ranged from 2 to 5 (mean: 2.4; median: 2) (Figure 1). Affymetrix U133A data on mRNA levels for EGFR were available in five of the amplified cases. Three of these (Table 2) showed increased EGFR mRNA levels greater than two-fold of the average EGFR mRNA level in EGFR-nonamplified tumors, and the remaining two tumors showed no significant increase above the average EGFR mRNA level. The mRNA data were not available in the other six EGFR-amplified tumors. No statistically significant correlation between gene copy number and level of EGFR transcript was found in this small number of amplified cases. Of the 164 tumors without EGFR gene amplification, mRNA data were available in 56 tumors. All but one tumor showed normal mRNA levels. The discordant case showed a 7.4-fold increase in mRNA level (data not shown).

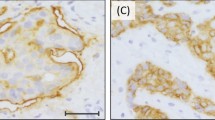
EGFR protein expression by immunohistochemistry and gene amplification by CISH. (a) 0 by immunohistochemistry, (b) 1+ by immunohistochemistry, (c) 2+ by immunohistochemistry, (d) 3+ by immunohistochemistry, (e) gene amplification (10–12 gene copies per nucleus) by CISH, (f) no gene amplification (2–3 gene copies per nucleus) by CISH.
By immunohistochemistry, the majority of breast carcinomas demonstrated 0–1+ immunoreactivity (162/175, 94%). Eight of the 11 breast carcinomas with amplified EGFR showed 3+ immunoreactivity, two tumors demonstrated 2+ and one tumor was scored as 1+ (Table 1). There was a strong correlation between 3+ immunoreactivity and gene amplification (P<0.0001, Fisher's exact test). Three of the 164 nonamplified tumors demonstrated EGFR protein overexpression. Two of these three tumors were poorly differentiated invasive ductal carcinomas and were 2+ by immunohistochemistry, the third tumor was an invasive pleomorphic lobular carcinoma and showed immunoreactivity of 3+ for EGFR without gene amplification.
Specific assays for the most frequent EGFR mutations in lung adenocarcinomas, exon 19 in-frame deletions and the exon 21 L858R point mutation, were used to analyze all EGFR-amplified tumors, and the one tumor with 3+ EGFR immunohistochemistry without EGFR gene amplification. None of the tumors showed either of these hotspot mutations in the EGFR gene (Table 2).
We evaluated the clinical and pathologic features of EGFR-amplified breast cancers in an effort to determine clinically relevant associations (Table 3). In all, 10 of these 11 tumors were poorly differentiated high-grade invasive ductal carcinoma, and one was a spindle cell metaplastic carcinoma with focal squamous differentiation. All of them were negative for estrogen receptor and progesterone receptor, but three of them were positive for HER-2 (Table 3). EGFR amplification appears to be inversely correlated with estrogen receptor expression. There was no correlation between EGFR amplification and HER-2 amplification. Three of the 11 patients developed distant metastases at 40, 42, and 48 months, respectively, after the initial diagnoses (Table 3). The first two patients (No. 6 and 7) died of disease at 84 and 55 months, respectively, and the third patient No. 9) is alive with lung and bone metastases at 89 months. One other patient (No. 8) died of unrelated causes at 34 months. The mean follow-up of the 11 patients is 73 months. Owing to the limited number of informative cases, we were unable to determine whether EGFR amplification and/or EGFR overexpression is an independent prognostic indicator.
Discussion
Although the EGFR gene was identified more than two decades ago,21 clinical interest in the gene has recently been heightened by the discovery of EGFR inhibitors. In 1996, Yang et al22 demonstrated that treatment with genistein, an inhibitor of tyrosine kinase activity, inhibited EGF-induced tyrosine phosphorylation and degradation of EGFR in HepG2 cells, suggesting that tyrosine kinase activity is required for either the internalization or the degradation of EGF–EGFR receptor complexes. The use of EGFR kinase inhibitors has recently received FDA approval for use in cancer therapy.
In this study, we used CISH to detect EGFR gene amplification in breast carcinomas. Our data revealed that EGFR gene amplification is an infrequent event in breast cancer, occurring in only 6% of tumors. This percentage is in the middle of the range reported by the few previous studies that have examined EGFR copy number in breast cancer (0.8–14%).23, 24
EGFR overexpression was seen in 6% tumors in our current study, which correlated well with gene amplification. Most studies that have reported a higher percentage of EGFR overexpression have not evaluated gene amplification.11, 12, 13 Differences in the prevalence of EGFR overexpression reported by different studies may be due to variations in techniques and type of antibodies used, criteria for determining overexpression and interobserver variability. For example, Harris et al11 measured EGFR in 221 primary breast cancers by ligand binding with 125I-labelled EGF, and high-affinity sites were quantitated. Tsutsui et al12 used a primary EGFR monoclonal antibody (Kyokutou Seiyaku, Tokyo, Japan) for assessing EGFR expression, and interpreted overexpression as ‘tumors exhibiting definite staining of the cancer cells’. In our current study, tumors with 1+ staining intensity were interpreted as negative for overexpression. Our stringent criteria in defining EGFR overexpression appeared to be the major contributing factor to the apparent low prevalence of EGFR overexpression among breast carcinomas in this study.
We found no correlation of EGFR amplification and HER-2 status. Of the 11 tumors showing EGFR gene amplification, three tumors (27%) showed HER-2 overexpression. These three tumors also showed HER-2 gene amplification. This proportion of HER-2 positivity approximates the expected percentage in breast cancers in general. The 11 EGFR-amplified tumors were uniformly estrogen receptor/progesterone receptor-negative, consistent with findings by other investigators.23
There are contradictory reports in the literature on the prognostic significance of EGFR overexpression and its relationship with known prognostic factors.25, 26, 27, 28 In the only study that examined the survival impact of EGFR gene amplification, no correlation was found.23 The clinical significance of EGFR amplification and/or EGFR overexpression could not be independently evaluated in our current study due to the small number of informative cases.
Low-level amplification of EGFR in concert with EGFR mutation is present in some lung adenocarcinoma cell lines29 and we (M Ladanyi, unpublished data) and others have also observed that many clinical lung cancer samples show evidence of copy number gains of the mutant allele.30 Based on these considerations, it was of interest to screen the EGFR-amplified tumors in the present study for the activating mutations in exon 19 and 21 that are commonly detected in lung cancers. However, no mutations were found.
EGFR gene amplification generally results in increased protein expression in breast carcinomas. Apparent EGFR protein overexpression without gene amplification occurred in only 2% of tumors in this study, and its mechanism needs to be further investigated. Overall, approximately 6% of breast carcinomas show moderate- to low-level EGFR amplification associated with genuine EGFR protein overexpression. A small minority of breast cancers could be responsive to EGFR-targeted therapy, and this carefully selected subset of patients should be considered for clinical trials evaluating EGFR antibodies or small inhibitory molecules.
References
Cohen S, Ushiro H, Stoscheck C, et al. A native 170,000 epidermal growth factor receptor-kinase complex from shed plasma membrane vesicles. J Biol Chem 1982;257:1523–1531.
McCune BK, Earp HS . The epidermal growth factor receptor tyrosine kinase in liver epithelial cells. The effect of ligand-dependent changes in cellular location. J Biol Chem 1989;264:15501–15507.
Alroy I, Yarden Y . The ErbB signaling network in embryogenesis and oncogenesis: signal diversification through combinatorial ligand-receptor interactions. FEBS Lett 1997;410:83–86.
Burgering BM, Coffer PJ . Protein kinase B (c-Akt) in phosphatidylinositol-3-OH kinase signal transduction. Nature 1995;376:599–602.
Liu W, Li J, Roth RA . Heregulin regulation of Akt/protein kinase B in breast cancer cells. Biochem Biophys Res Commun 1999;261:897–903.
Muthuswamy SK, Gilman M, Brugge JS . Controlled dimerization of ErbB receptors provides evidence for differential signaling by homo- and heterodimers. Mol Cell Biol 1999;19:6845–6857.
Chan TO, Rittenhouse SE, Tsichlis PN . AKT/PKB and other D3 phosphoinositide-regulated kinases: kinase activation by phosphoinositide-dependent phosphorylation. Annu Rev Biochem 1999;68:965–1014.
Lynch TJ, Bell DW, Sordella R, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med 2004;350:2129–2139.
Paez JG, Janne PA, Lee JC, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science 2004;304:1497–1500.
Pao W, Miller V, Zakowski M, et al. EGF receptor gene mutations are common in lung cancers from ‘never smokers’ and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc Natl Acad Sci USA 2004;101:13306–13311.
Harris AL, Nicholson S, Sainsbury JR, et al. Epidermal growth factor receptors in breast cancer: association with early relapse and death, poor response to hormones and interactions with neu. J Steroid Biochem 1989;34:123–131.
Tsutsui S, Kataoka A, Ohno S, et al. Prognostic and predictive value of epidermal growth factor receptor in recurrent breast cancer. Clin Cancer Res 2002;8:3454–3460.
Walker RA, Dearing SJ . Expression of epidermal growth factor receptor mRNA and protein in primary breast carcinomas. Breast Cancer Res Treat 1999;53:167–176.
Kallioniemi OP, Wagner U, Kononen J, et al. Tissue microarray technology for high-throughput molecular profiling of cancer. Hum Mol Genet 2001;10:657–662.
Kononen J, Bubendorf L, Kallioniemi A, et al. Tissue microarrays for high-throughput molecular profiling of tumor specimens. Nat Med 1998;4:844–847.
Skacel M, Skilton B, Pettay JD, et al. Tissue microarrays: a powerful tool for high-throughput analysis of clinical specimens: a review of the method with validation data. Appl Immunohistochem Mol Morphol 2002;10:1–6.
Bhargava R, Lal P, Chen B . Chromogenic in situ hybridization for the detection of HER-2/neu gene amplification in breast cancer with an emphasis on tumors with borderline or low-level amplification. Does it measure up to fluorescent in situ hybridization? Am J Clin Pathol 2005;123:237–243.
Bhargava R, Lal P, Chen B . Feasibility of using tissue microarrays for the assessment of HER-2 gene amplification by fluorescence in situ hybridization in breast carcinoma. Diagn Mol Pathol 2004;13:213–216.
LaTulippe E, Satagopan J, Smith A, et al. Comprehensive gene expression analysis of prostate cancer reveals distinct transcriptional programs associated with metastatic disease. Cancer Res 2002;62:4499–4506.
Pan Q, Pao W, Ladanyi M . Rapid PCR-based detection of epidermal growth factor receptor gene mutations in lung adenocarcinomas. J Mol Diagn (in press).
Shimizu N, Behzadian MA, Shimizu Y . Genetics of cell surface receptors for bioactive polypeptides: binding of epidermal growth factor is associated with the presence of human chromosome 7 in human-mouse cell hybrids. Proc Natl Acad Sci USA 1980;77:3600–3604.
Yang EB, Wang DF, Mack P, et al. Genistein, a tyrosine kinase inhibitor, reduces EGF-induced EGF receptor internalization and degradation in human hepatoma HepG2 cells. Biochem Biophys Res Commun 1996;224:309–317.
Al Kuraya K, Schraml P, Torhorst J, et al. Prognostic relevance of gene amplifications and coamplifications in breast cancer. Cancer Res 2004;64:8534–8540.
Ro J, North SM, Gallick GE, et al. Amplified and overexpressed epidermal growth factor receptor gene in uncultured primary human breast carcinoma. Cancer Res 1988;48:161–164.
Cerra M, Cecco L, Montella M, et al. Epidermal growth factor receptor in human breast cancer comparison with steroid receptors and other prognostic factors. Int J Biol Markers 1995;10:136–142.
Fox SB, Smith K, Hollyer J, et al. The epidermal growth factor receptor as a prognostic marker: results of 370 patients and review of 3009 patients. Breast Cancer Res Treat 1994;29:41–49.
Pirinen R, Lipponen P, Syrjanen K . Expression of epidermal growth factor receptor (EGFR) in breast cancer as related to clinical, prognostic and cytometric factors. Anticancer Res 1995;15:2835–2840.
Toi M, Nakamura T, Mukaida H, et al. Relationship between epidermal growth factor receptor status and various prognostic factors in human breast cancer. Cancer 1990;65:1980–1984.
Amann J, Kalyankrishna S, Massion PP, et al. Aberrant epidermal growth factor receptor signaling and enhanced sensitivity to EGFR inhibitors in lung cancer. Cancer Res 2005;65:226–235.
Kosaka T, Yatabe Y, Endoh H, et al. Mutations of the epidermal growth factor receptor gene in lung cancer: biological and clinical implications. Cancer Res 2004;64:8919–8923.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Bhargava, R., Gerald, W., Li, A. et al. EGFR gene amplification in breast cancer: correlation with epidermal growth factor receptor mRNA and protein expression and HER-2 status and absence of EGFR-activating mutations. Mod Pathol 18, 1027–1033 (2005). https://doi.org/10.1038/modpathol.3800438
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.3800438
Keywords
This article is cited by
-
Endosomal protein expression of γ1-adaptin is associated with tumor growth activity and relapse-free survival in breast cancer
Breast Cancer (2024)
-
Breast cancer with scalp metastases: a case report
Journal of Medical Case Reports (2023)
-
A SERS/fluorescence dual-mode immuno-nanoprobe for investigating two anti-diabetic drugs on EGFR expressions
Microchimica Acta (2023)
-
A triple-negative breast cancer surrogate subtype classification that correlates with gene expression subtypes
Breast Cancer Research and Treatment (2022)
-
Rac inhibition as a novel therapeutic strategy for EGFR/HER2 targeted therapy resistant breast cancer
BMC Cancer (2021)
