
Abstract
The tumor suppressor gene PTEN/MMAC1 is located on chromosome 10q23.3. Inactivation of PTEN, either by mutations, deletions, or promoter hypermethylation, has been identified in a wide variety of tumors. Inactivation of the two alleles of PTEN is required, because it is a tumor suppressor gene. Immunohistochemical staining may be an effective screening method to demonstrate the absence of the protein in tumors exhibiting PTEN inactivation. We studied a tissue microarray, constructed from paraffin-embedded blocks of 95 endometrial carcinomas, 38 of them previously evaluated for alterations in PTEN. We also studied cell blocks obtained from one PTEN-defective endometrial cancer cell line, after transfection with either a plasmid encoding wild-type PTEN or the empty vector. The tumor samples were tested with four different anti-PTEN commercial antibodies: a polyclonal antibody, the monoclonal antibody 28H6, the monoclonal antibody 10P03, and the monoclonal antibody 6.H2.1. Results were correlated with the presence of abnormalities in PTEN, as well as with the immunohistochemical expression of phosphorylated AKT. Antibody 28H6 produced a predominant nuclear staining, while the other three antibodies produced a predominant cytoplasmic staining. There was no significant correlation between the results obtained with the four antibodies. The monoclonal antibody 6.H2.1 was the only one that exhibited a correlation with the presence of molecular alterations in PTEN, and a statistically significant association with immunostaining for phosphorylated AKT (r=−0.249, P=0.037). The monoclonal antibody 10P03 exhibited an association with phospho-AKT that did not have statistical significance. Both 6.H2.1 and 10P03 antibodies stained PTEN-transfected cells, and were negative in the PTEN-deficient cell line blocks. The polyclonal antibody and the monoclonal antibody 28H6 produced positive staining in PTEN-deficient cell line blocks, suggesting nonspecific staining. The results indicate that monoclonal antibody 6.H2.1 may be a suitable alternative for tumors with inactivation of PTEN.
Similar content being viewed by others
Main
The tumor suppressor gene PTEN1 is located on chromosome 10q23. It is somatically mutated or deleted in several types of tumors.2, 3 PTEN encodes a 403 amino-acid dual specificity phosphatase containing a region of homology to tensin and auxilin, which are two cytoskeletal proteins. Among other activities, PTEN antagonizes the PI3K/AKT pathway by dephosphorylating PIP3, resulting in a decreased translocation of AKT to cellular membranes, and subsequent downregulation of AKT phosphorylation and activation.4, 5 The PI3K/AKT pathway plays a key role in the regulation of cellular homeostasis. Activation of cell surface receptors recruits PI3-K to the receptor where it phosphorylates the PtdIns(4,5)P2 substrate to generate PtdIns(3,4,5)P3, which is recognized by the protein kinase AKT and its regulator PDK1. In the cell membrane, AKT is phosphrylated in serine and threonine residues. Activated AKT modulates the expression of several genes involved in suppression of apoptosis and cell cycle progression.
PTEN has been implicated in an important number of human tumors of different sites, such as the thyroid, endometrium, brain, prostate, or in melanomas.1 Inactivation of the two alleles of PTEN, by mutation or deletion, is required because it is a tumor suppressor gene. Since the frequency of PTEN suppression in some tumors exceeds those of PTEN mutations or deletions, it has been suggested that promoter hypermethylation may also account for its inactivation in a subset of tumors.6, 7
Several studies have suggested that the immunohistochemical evaluation of PTEN expression may be an effective screening method that can demonstrate the absence of the protein in a lesion, whatever the underlying mechanism.8 However, some variability has been observed with different antibodies and techniques,9, 10, 11 particularly when correlating the immunohistochemical results with the presence of molecular alterations.
In the present study, we tested the immunohistochemical performance characteristics of four anti-PTEN antibodies that are commercially available on a tissue microarray of 95 endometrial carcinomas, 38 of them previously characterized at the molecular level for mutations,12 loss of heterozygosity, and promoter hypermethylation of PTEN.13 Immunohistochemical results were correlated with abnormalities in the gene, and with immunostaining of phosphorylated AKT (phospho-AKT). Cell blocks from the PTEN-deficient, Ishikawa 3-H-12 cell line after transfection with either a plasmid encoding wild-type PTEN or the empty vector were also stained. The main goal of the study was to ascertain the best immunohistochemical approach to assess the presence of abnormalities in PTEN.
Materials and methods
Case Selection and Tissue Microarray Design
A tissue microarray was constructed from paraffin-embedded blocks of 95 endometrial carcinomas. They were obtained from the Department of Pathology of Hospital Santa Creu i Sant Pau, Barcelona, Spain. The specimens were collected between 1996 and 2001. They included: 25 endometrioid carcinomas grade I, 34 endometrioid carcinomas grade II, 19 endometrioid carcinomas grade III, 10 serous carcinomas, four clear cell carcinomas, and the epithelial components of three malignant mixed Müllerian tumors.
A Tissue arrayer device (Beecher Instrument, MD, USA) was used. All endometrial carcinomas were histologically reviewed and representative tumor areas were marked in the corresponding paraffin blocks. Two selected cylinders (0.6 mm in largest diameter) from two different tumor areas were included for each case. Normal control tissues from the same specimens were also included. Thus, two different tissue microarray blocks were constructed; each of them contained 186 cylinders.
Of the 95 endometrial carcinomas, 38 had been previously evaluated for mutations in PTEN,12 as well as for loss of heterozygosity analysis at 10q23, and promoter hypermethylation.13 PTEN mutations were detected in 17 of the 38 tumors (44.7%); missense mutations in four patients; and frameshift mutations in the remaining 13 cases. Two tumors exhibited two different mutations, and all four were missense mutations. Loss of heterozygosity was analyzed by microsatellite PCR in four different loci, and it was detected at one or more loci in nine tumors (23.6%). Methylation-specific PCR demonstrated PTEN promoter hypermethylation in five endometrial carcinomas (13%). Overall, 22 of the 38 endometrial carcinomas (57.8%) presented at least one type of PTEN abnormality. Moreover, the two hits required for the inactivation of a tumor suppressor gene (coexistence of somatic mutation, and/or hypermethylation, and/or loss of heterozygosity) occurred in 10 endometrial carcinomas (26%).
The Ishikawa 3-H-12 cell line was obtained from the American Type Culture Collection (Manassas, VA, USA). Ishikawa 3-H-12 cells14, 15 contain two frameshift mutations in PTEN, a deletion of one base pair at codon 289, and a deletion of four base pairs at codons 317–318. These mutations result in a truncated protein that lacks the carboxy-terminal region. Cells were grown in Dulbeco's modified Eagle's Medium supplemented with 10% fetal bovine serum, 1 mM HEPES, 1 mM sodium pyruvate, 2 mM L-glutamine and penicillin/streptomycin. For transfection, Ishikawa 3-H-12 cells were plated in M24-well dishes and transfected with a plasmid containing cDNA for PTEN (PRK5-PTEN) or the empty vector (PRK5) using polyethylenimide reagent. Separate samples containing cells transfected with the empty vector or the plasmid encoding for PTEN were harvested by trypsinization and embedded in 1% agarose in phosphate-buffered saline, fixed in formalin, and embedded in paraffin.
Immunohistochemistry
Tissue microarray blocks, and paraffin-embedded blocks of Ishikawa 3-H-12 cells transfected with PRK5 or PRK5-PTEN were sectioned at a thickness of 3 μm, and dried for 16 h at 56°C. They were dewaxed in xylene, rehydrated through a graded ethanol series, and washed with phosphate-buffered saline. Antigen retrieval was achieved by heat treatment in a pressure-cooker for 2 min in 10 mM citrate buffer (pH 6.5) or EDTA buffer (pH 8.0) (Table 1). Endogenous peroxidase was blocked. The antibodies used are described in Table 1. Four different anti-PTEN commercial antibodies were used: polyclonal antibody from Zymed, monoclonal antibody 28H6 from Novocastra, monoclonal antibody 10P03 from Neomarkers, and monoclonal antibody 6H2.1 from Cascade Bioscience. All four antibodies were made to react against epitopes located in the carboxy-terminal region of PTEN. An antiphosphorylated-AKT polyclonal antibody from Cell Signalling was also used. The sections were incubated during 30 min with the polyclonal antibody, the monoclonal antibody 28H6, and the antiphosphorylated-AKT antibody. The 10P03 antibody and the 6H2.1 were incubated overnight at 4°C. The reaction was visualized with the EnVision Detection Kit (DAKO, Glostrup, Denmark) using diaminobenzidine chromogen as substrate. Sections were counterstained with Harris hematoxylin. The immunohistochemical reactions were performed in a Dako Autostainer LV-1 immunostainer. Endometrial stroma was used as a positive control and the negative control was run without addition of the primary antibody. Antigen preservation was verified with vimentin and Ki-67 immunostaining. Only samples exhibiting positive vimentin and Ki-67 staining, as well as optimal internal control were evaluated.
Immunohistochemical results were evaluated by two different pathologists (XM-G, and JP). Uniform pre-established criteria were used. Immunoreactivity was graded semiquantitatively by considering the percentage and intensity of the staining of the tumor cells. A histological score was obtained from each sample, which ranged from 0 (no immunoreaction) to 300 (maximum immunoreactivity). The score was obtained by applying the following formula: Hscore=1 × (% light staining)+2 × (% moderate staining)+3 × (% strong staining). The Hscore formula was applied for the polyclonal antibody, and the 10P03, and 6H2.1 monoclonal antibodies. Immunostaining for 28H6 was assessed by considering the percentage of positive cells because the positivity was homogeneous in each tumor sample. Since each tissue microarray included two different tumor cylinders from each case, immunohistochemical evaluation was performed after examining both samples.
Validation of the Tissue Microarray
The reproducibility of tissue microarray immunostaining was confirmed by comparing tissue microarray results with those obtained in sections from the corresponding paraffin blocks of 37 randomly selected cases. The overall concordance was 89.2%. The Kappa index of agreement between the two methods ranged from 0.68 to 0.83.
Statistical Analysis
The Wilcoxon rank sum was used to assess association between quantitative and dichotomous variables. The Spearman rank correlation coefficient was used to assess association between quantitative variables.
Results
From the 95 cases of the tissue microarray, two cases were excluded because there was no representative tumor sample. The number of cylinders missed in the construction of the tissue microarray varied from two to fifteen cases. Six cases were excluded because unreliable antigen preservation verified with vimentin and Ki67 immunostaining. Results are shown in Table 2.
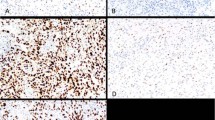
The 10P03 antibody was evaluated in 76 cases. A total of 11 cases were missed in the construction of the tissue microarray and two cases had no representative tumor. Cytoplasmic staining was found in 55 cases (72%) and varied from an Hscore of 10 to 290 (mean=58). Nuclear positive staining was not observed (Figure 1).

Immunostaining pattern of an endometrial carcinoma sample with antibody 10P03.
The 28H6 antibody was evaluated in 80 cases. Seven cases were missed and two cases had no representative tumor sample. Predominant nuclear staining was seen in 63 cases (79%) (Figure 2).

Immunohistochemical pattern obtained in one tumor with antibody 28H6. Notice strong nuclear staining.
The polyclonal antibody was evaluated in 82 cases. Five cases were missed in the tissue microarray construction and two cases had no representative tumor. All cases showed cytoplasmic staining (100%), and the intensity varied from low to high (mean=130; min. 10, max. 300) (Figure 3).

Staining pattern found in an endometrial carcinoma with the polyclonal antibody.
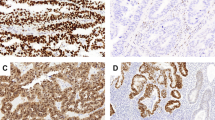
The 6H2.1 antibody was evaluated in 72 cases. In all, 15 cylinders were missed in the tissue microarray construction and two cases had no representative tumor. Positive cytoplasmic staining was noted in 50 cases (69%) and the intensity varied from an Hscore of 5 to 290 (mean: 46) (Figure 4).

Immunohistochemical patterns obtained with antibody 6H2.1 in two cases. Strong and diffuse positivity (a). Focal positivity in tumor cells in comparison with strong positivity in normal stromal cells (b).
Overall, there was no significant correlation between the results obtained with the four antibodies. However, it is worth mentioning that the monoclonal antibody 6H2.1 had a significant negative correlation with the polyclonal antibody (r=−0.337; P=0.005).
Phospho-AKT was evaluated in 83 cases. Three cases were missed and three cases had no tumor left for evaluation. Granular cytoplasmic staining was seen in 40 cases (48%) (mean=20; min. 0, max. 235) (Figure 5). There was a statistically significant negative correlation between the immunoexpression of PTEN with the 6H2.1 antibody and phospho-AKT (r=−0.249) (P=0.037). Cases exhibiting low PTEN staining with the 6H2.1 antibody showed an increase in phospho-AKT immunostaining. The monoclonal antibody 10P03 exhibited an association with phospho-AKT, which did not have statistical significance (r=−0.05) (P=0.388). There was no negative correlation between PTEN immunostaining assessed either by the polyclonal antibody (r=0.379) (P=0.0005) or the monoclonal antibody 28H6 and the presence of positive immunostaining for phospho-AKT.

Positive immunostaining for phosphorylated AKT in one tumor.
There was no statistically significant correlation between the presence of molecular alterations of PTEN (mutations, deletions, and promoter hypermethylation) and the immunohistochemical results with any of the four antibodies (see Table 3). However, the 6H2.1 antibody was the only one that showed lower immunostaining in the tumors that exhibited PTEN molecular alterations (mean Hscore 43) in comparison with those with normal PTEN (mean Hscore 65.5) (Table 3). For the remaining antibodies, the mean Hscore (polyclonal antibody and 10P03) was higher in tumors with molecular alterations of PTEN in comparison with those showing normal PTEN. Moreover, the percentage of positive cells for the monoclonal antibody 28H6 in the tumors with molecular alterations in PTEN was very high (80%).
Both 6.H2.1 and 10P03 antibodies stained PTEN transfected, Ishikawa 3-H-12 cells, and were negative in the PTEN-deficient Ishikawa 3-H-12 cell line blocks (Figures 6 and 7). The polyclonal antibody, and the monoclonal antibody 28H6 produced positive staining in both PTEN-deficient and PTEN transfected, Ishikawa 3-H-12 cell line blocks, suggesting nonspecific staining.

Staining pattern obtained for antibodies 10P03 (a), 28H6 (b), polyclonal antibody (c), and 6H2.1 antibody in the Ishikawa 3-H-12 cells transfected with a plasmid containing cDNA for PTEN (PRK5-PTEN). Notice the presence of the expected positive staining with each of the four antibodies.

Staining pattern obtained for antibodies 10P03 (a), 28H6 (b), polyclonal antibody (c), and 6H2.1 antibody in the Ishikawa 3-H-12 cells transfected with the empty vector (PRK5). Notice the presence of unexpected positive staining with the 28H6 monoclonal antibody, and the polyclonal antibody.
Finally, there was no statistical association between PTEN or AKT-P expression and the clinical and pathological data, including histological grade.
Discussion
The tumor suppressor gene named PTEN (phosphatase and tensin homologue deleted from chromosome 10),1 also called MMAC1 (mutated in multiple advanced cancers), is located on chromosome 10q23. 3.1, 2 Its amino-acid sequence indicates that the protein resembles two different types of proteins: tyrosine phosphatases, which are enzymes that remove phosphate groups from the amino acid tyrosine in other proteins, and tensin, a protein that helps connecting the cell's internal skeleton of protein filaments to its external environment. PTEN antagonizes the PI3K/AKT pathway by dephosphorylating PIP3, which results in a decreased translocation of AKT to cellular membranes, and subsequent downregulation of AKT phosphorylation activation.4, 5 Thus, decreased expression of PTEN leads to increased levels of phospho-AKT, which results in an activation of antiapoptotic proteins (BAD), inactivation of members of the forkhead family of transcription factors (FKHRL1) as well as inactivation of proteins involved in cell cycle progression (p27 and p21). Both suppression of apoptosis and induction of cell cycle are processes involved in cancer development.
PTEN may be inactivated by several mechanisms. PTEN is mutated in the germline of patients with a rare autosomal dominant cancer syndrome called Cowden's disease, and its variants.16 However, PTEN is also frequently somatically mutated in tumors from various tissues.2, 3 Germline and somatic mutations in PTEN occur mostly in the phosphatase domain, between residues 122 and 132, in exon 5. Altogether, 75% of the germline mutations correspond to nonsense (36%), frameshift (27%), and splice-site mutations (11%), which result in a truncated protein, while missense point mutations represent the remaining 25%. The spectrum of somatic mutations shows characteristic features for the tumors of different organs. For example, in endometrial carcinoma, 60% of the mutations are frameshift, while they account for 37% in glioblastomas.2, 3 PTEN may be also inactivated by deletion, as shown by the elevated frequency of loss of heterozygosity in different tumor types.17, 18 Finally, a third proposed mechanism for PTEN inactivation is promoter hypermethylation.6, 7 However, the true significance of PTEN promoter methylation is under discussion.19, 20 Some controversy exists in the literature regarding the possible interference of a processed PTEN pseudogene (psiPTEN) with PTEN. psiPTEN is located on chromosome 9, and the genomic sequence has a 1.2 kb region with 98% homology to the entire coding region of PTEN.20 The homology region includes the promoter. psiPTEN may also interfere at the mRNA level in molecular studies, because the mRNA sequence of these genes shares a high degee of homology. Despite the possible interference with mRNA, no evidence of translation to protein of the psiPTEN mRNA in endometrial carcinoma has been reported so far. Since the frequency of PTEN suppression in some types of tumors exceeds that of PTEN mutations or deletions, it is very likely that epigenetic mechanisms, like promoter hypermethylation, may account for its inactivation in some cases.
PTEN is frequently abnormal in endometrial carcinomas. Loss of heterozygosity at chromosome 10q23 occurs in 40% of cases.21, 22 Moreover, somatic PTEN mutations are also common in endometrial carcinomas, and they are almost exclusively restricted to endometrioid endometrial carcinomas, occurring in 37–61% of them.12, 23, 24, 25 Interestingly, PTEN mutations have been detected in endometrial hyperplasias with and without atypia (19 and 21%, respectively), both of them currently regarded as precursor lesions of endometrioid endometrial carcinomas.26 Even more, identical PTEN mutations have been detected in hyperplasias coexisting with endometrioid endometrial carcinomas, which suggests that PTEN mutations are early events in the development of endometrioid endometrial carcinoma.
The hypothesis that loss of functional PTEN could be identified by immunohistochemistry has led to the suggestion that PTEN immunostaining may be an effective method to screen for abnormal PTEN expression in tumors and premalignant lesions.8 At this regard, the important role of PTEN in endometrial tumorigenesis has prompted some authors to suggest that the immunohistochemical demonstration of decreased PTEN expression or function in endometrial biopsies may be clinically relevant to identify premalignant lesions that are likely to progress to endometrial carcinoma. Thus, PTEN immunostaining could be an important pathologic tool to assess the presence of precancerous clones in endometrial hyperplasias.8
Immunohistochemistry has become an indispensable tool in diagnostic and experimental pathology. However, it is important to standardize immunohistochemical techniques to avoid interlaboratory variations in staining and interpretation, which could have an impact in the management of patients. Obviously, the selection of the most appropriate antibody is crucial to obtain reliable and reproducible results. Some studies have assessed the specificity of staining of other antibodies used in immunohistochemistry.27 In addition to the specificity of the antibody. It is important to keep in mind that the quality of immunostaining depends also on other factors such as tissue fixation and processing, unmasking of epitopes, and sensitivity of the detection systems.
In the present study, we assessed the immunohistochemical performance characteristics of four anti-PTEN antibodies that are commercially available. The immunohistochemical results were correlated with the presence of mutations, deletions, and promoter hypermethylation of PTEN in 38 cases. Since PTEN acts as an antagonist of PI3-kinase-dependent signalling pathway, the results were also correlated with positivity for phosphoAKT.
We observed a wide variability in the results obtained with the four antibodies, and the concordance between them was poor. Three of the antibodies demonstrated a positive cytoplasmic staining (the polyclonal antibody, and the monoclonal antibodies 10P03 and 6.H2.1), while the monoclonal antibody 28H6 showed a predominant positive nuclear staining.
Since it is well known that decreased expression of PTEN leads to increased levels of phospho-AKT, PTEN immunostaining with each of these four antibodies was correlated with immunoreaction for phospho-AKT, which specifically recognizes phosphorylation on serine 473 of the Akt. The specificity of phospho-Akt antibody has been previously demonstrated in mice with deletion of PTEN.28 Immunoreactivity for phospho-AKT was obtained in 48% of the cases. Positive phospho-AKT immunostaining exhibited a statistically significant association with decreased staining with the monoclonal antibody 6.H2.1. It also showed an association with negative immunostaining with the monoclonal antibody 10P03, but the results did not show statistical significance. Results obtained with the polyclonal antibody, and the monoclonal antibody 28H6 did not correlate with phospho-AKT immunostaining.
Immunohistochemical results were also correlated with the presence of molecular alterations of PTEN (mutations, loss of heterozygosity, or promoter hypermethylation) in 38 cases. The results obtained with the four tested antibodies did not get statistically significant associations with these molecular alterations. However, the 6H2.1 antibody was the only one that showed lower immunostaining in the tumors that exhibited PTEN alterations in comparison with those with normal PTEN. The other antibodies showed unexpected results. For example, the mean Hscores obtained with the polyclonal antibody and the 10P03 monoclonal antibody were higher in tumors with molecular alterations of PTEN in comparison with those showing normal PTEN. Moreover, the monoclonal antibody 28H6 produced a strong positive nuclear pattern in the vast majority (80%) of tumors with molecular alterations of PTEN, which are supposed to exhibit decreased PTEN protein expression or function.
To provide further evidence of the specificity of the four antibodies, we performed immunohistochemical analysis on paraffin-embedded cell blocks, obtained from the PTEN-defective Ishikawa 3-H-12 endometrial cancer cell line transfected with a vector encoding wild-type PTEN or the control empty vector. Both 6.H2.1 and 10P03 antibodies stained PTEN-transfected cells, and were negative in the PTEN-deficient cell line blocks. The polyclonal antibody and the monoclonal antibody 28H6 produced positive staining in PTEN-deficient cell line blocks, suggesting nonspecific staining.
Taken together, our results demonstrate that the polyclonal antibody and the monoclonal antibody 28H6 display nonspecific staining. The monoclonal antibody 6.H2.1 is the only antibody that correlated statistically with phospho-AKT, and also the only one to show correlation with the presence of molecular alterations in PTEN (mutations, deletions or promoter hypermethylation). These data indicate that the 6.H2.1 antibody is a suitable alternative for tumors with inactivation of PTEN.
References
Li J, Yen C, Liaw D, et al. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast and prostate cancer. Science 1997;275:1943–1946.
Bonneau D, Longy M . Mutations of the human PTEN gene. Hum Mutat 2000;16:109–122.
Unnisa I, Schriml LM, Dean M . Mutational spectra of PTEN/MMAC1 gene: a tumor suppressor with lipid phosphatase activity. J Natl Cancer Inst 1999;91:1922–1932.
Downes CP, Bennett D, McConnachie G, et al. Antagonism of PI3-kinase-dependent signalling pathways by the tumour supressor protein, PTEN. Biochem Soc Trans 2001;29:846–851.
Maehama T, Dixon JE . PTEN. a tumour suppressor that functions as a phospholipids phosphatase. Trends Cell Biol 1999;9:125–128.
Soria JC, Lee HY, Lee JI, et al. Lack of PTEN expression in non-small cell lung cancer could be related to promoter methylation. Clin Cancer Res 2002;8:1178–1184.
Zhou XP, Gimm O, Hampel H, et al. Epigenetic PTEN silencing in malignant melanomas without PTEN mutation. Am J Pathol 2000;157:1123–1128.
George L, Mutter, Ming-Chieh Lin, Jeffrey T, et al. Altered PTEN expression as a diagnostic marker for the earliest endometrial precancers. J Natl Cancer Inst 2000;92:924–931.
Terakawa N, Kanamori Y, Yoshida S . Loss of PTEN expression followed by Akt phosphorylation is a poor prognostic factor for patients with endometrial cancer. Endocr Relat Cancer 2003;10:203–208.
Salvesen Hb, Stefansson I, Kalvenes MB, et al. Loss of PTEN expression is associated with metastatic disease in patients with endometrial carcinoma. Cancer 2002;94:2185–2191.
Kanamori Y, Kigawa J, Itamochi H, et al. PTEN expression is associated with prognosis for patients with advanced endometrial carcinoma undergoing postoperative chemotherapy. Int J Cancer 2002;100:686–689.
Bussaglia E, del Rio E, Matias-Guiu X, et al. PTEN mutations in endometrial carcinomas. A molecular and clinicopathologic analysis of 38 cases. Hum Pathol 2000;31:312–317.
Bussaglia E, Pallares J, Matias-Guiu X, et al. LOH and promoter hypermethylation of PTEN in endometrial carcinoma. Int J Gynecol Pathol (submitted).
Nishida M, Kasahara K, Kaneko M, et al. Establishment of a new human endometrial adenocarcinoma cell line. Ishikawa cells. Containing estrogen and progesterone receptors. Nippon Sanka Fujinka Gakkai Zasshi 1985;37:1103–1111.
Nishida M, Kasahara K, Oki A, et al. Establishment of eighteen clones of Ishikawa cells. Hum Cell 1996;9:109–116.
Bussaglia E, Pujol RM, Gil MJ, et al. PTEN mutations in eight spanish families and one brazilian family with Cowden syndrome. J Invest Dermatol 2002;118:639–644.
Rasheed BK, McLendon RE, Friedmen HS, et al. Chromosome 10 deletion mapping in human gliomas: a common deletion region in 10q25. Oncogene 1995;10:2243–2246.
Gray IC, Philips SMA, Lee SJ, et al. Loss of the chromosomal region 10q23-25 in prostate cancer. Cancer Res 1995;55:4800–4803.
Salvesen HB, MacDonald N, Ryan A, et al. PTEN methylation is associated with advanced stage and microsatellite instability in endometrial carcinoma. Int J Cancer 2001;91:22–26.
Zysman MA, Chapman WB, Bapat B . Considerations when analyzing the methylation status of PTEN tumor supressor gene. Am J Pathol 2002;160:795–800.
Nagase S, Sato S, Tezuka F, et al. Deletion mapping on chromosome 10q25-q26 in human endometrial cancer. Br J Cancer 1996;74:1979–1983.
Peiffer SL, Herzog TJ, Tribune DJ, et al. Allelic loss of sequences from the long arm of chromosome 10 and replication errors in endometrial cancers. Cancer Res 1995;55:1922–1926.
Tashiro H, Blazes MS, Wu R, et al. Mutations in PTEN are frequent in endometrial carcinoma but rare in other common gynecological malignancies. Cancer Res 1997;57:3935–3940.
Kong D, Suzuki A, Zou TT, et al. PTEN1 is frequently mutated in primary endometrial carcinomas. Nat Genet 1997;17:143–144.
Risinger JI, Hayes AK, Berchuck A, et al. PTEN/MMAC1 mutations in endometrial cancers. Cancer Res 1997;57:4736–4738.
Levine RL, Cargile CB, Blazes MS, et al. PTEN mutations and microsatellite instability in complex atypical hyperplasia, a precursor lesion to uterine endometrioid carcinoma. Cancer Res 1998;58:3254–3258.
Van der loos CM, Houtkamp MA, de Boer OJ, et al. Immunohistochemical detection of interferon-γ fake or fact. J Histochem Cytochem 2001;49:699–710.
Wang S, Gao J, Lei Q, Rozengurt N, et al. Prostate-specific deletion of the murine PTEN tumor suppressor gene leads to metastasic prostate carncer. Cancer Cell 2003;4:209–221.
Acknowledgements
This work was supported by Grants FISS 01/1656, FISS PI020227, SAF 2004-05250, and 2002XT00115. JP holds a research fellowship from the Fondo de Investigaciones Sanitarias, Ministerio de Sanidad y Consumo (BEFI-02/9007)). JLMG and XD hold a postdoctoral fellowship supported by the Department d’Universitats, Recerca i Societat de la Informació, Generalitat de Catalunya (2002RED005 and, 2003RED0018). We thank Mrs S Mancilla, Ll Barrera, L Barragan, and A Salla for their technical assistance.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Pallares, J., Bussaglia, E., Martínez-Guitarte, J. et al. Immunohistochemical analysis of PTEN in endometrial carcinoma: a tissue microarray study with a comparison of four commercial antibodies in correlation with molecular abnormalities. Mod Pathol 18, 719–727 (2005). https://doi.org/10.1038/modpathol.3800347
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.3800347
Keywords
This article is cited by
-
Precise definition of PTEN C-terminal epitopes and its implications in clinical oncology
npj Precision Oncology (2019)
-
Molecular expression characteristics confirm the malignancy concealed by morphological alterations in endometrial cancer after fertility-preserving treatment
Archives of Gynecology and Obstetrics (2019)
-
Are the uterine serous carcinomas underdiagnosed? Histomorphologic and immunohistochemical correlates and clinical follow up in high-grade endometrial carcinomas initially diagnosed as high-grade endometrioid carcinoma
Modern Pathology (2018)
-
Lung neuroendocrine tumors: correlation of ubiquitinylation and sumoylation with nucleo-cytosolic partitioning of PTEN
BMC Cancer (2015)
