
Abstract
The Wnt signaling pathway is essential for normal development and organogenesis. However, inappropriate activation of Wnt signaling, which results in the nuclear translocation of β-catenin, is associated with the development of various types of neoplasm. In this study, we investigated possible mutations in the genes for components of this pathway, namely, CTNNB1 (the gene for β-catenin), AXIN1, and APC, in adenoid cystic carcinoma, by PCR, analysis of single-strand conformational polymorphism, and sequencing. Among a total of 20 cases of adenoid cystic carcinoma, seven cases (35%) were associated with mutations in one or more of these three components. A mutation in CTNNB1 was detected in one case. Five cases, including the case with a mutation in CTNNB1, were associated with missense mutations in AXIN1. An aberration in the mutation cluster region of APC was detected in two cases. Mutations trended to be detected more frequently in adenoid cystic carcinoma with solid growth pattern than that with tubular and cribriform growth pattern. In the cases in which we detected mutations, it is possible that the presence of the abnormal products of the mutated genes resulted in the inappropriate activation of the Wnt signaling pathway to tumorigenesis and the growth of adenoid cystic carcinoma.
Similar content being viewed by others

Main
The Wnt proteins form a large family of secreted signaling proteins, which have been found in organisms from the nematode to mammals. They have been implicated in the proliferation, morpho-logy, motility, and fate of cells, in axis formation in xenopus, and in organ development.1, 2 To date, roles for Wnt have been recognized in three distinct pathways: the canonical β-catenin pathway; the planar cell polarity pathway;3 and the Wnt Ca2+ pathway.4
The canonical β-catenin pathway has been investigated in greater detail than the other two pathways and it includes the adenomatous polyposis coli protein (apc), axin1 (the product of the AXIN1 gene), and β-catenin. When Wnt binds to a receptor, which consists of the Frizzled protein and LRP5/6,5 phosphorylation of the Disheveled protein occurs and results in inactivation of glycogen synthase kinase 3β (GSK3β). Since GSK3β phosphorylates β-catenin in the axin1–apc complex,6, 7 inactivation of GSK3β leads to a reduction in the extent of phosphorylation of β-catenin. Unphosphorylated β-catenin becomes dissociated from the axin1–apc complex and escapes degradation by the proteasome after ubiquitination.8 When the Frizzled protein is not activated by Wnt, β-catenin in the axin1–apc complex is phosphorylated by GSK3β, with subsequent degradation of β-catenin. Thus, Wnt increases the stability of β-catenin, by inactivating GSK3β, and, as a consequence, the cytoplasmic level of β-catenin rises. Then β-catenin in the cytoplasm is translocated to the nucleus, where it binds to the TCF/LEF-1 protein and enhances the transcriptional activity of the TCF/LEF-1 family. In this way, acting as a transcription factor, β-catenin stimulates the expression of various genes, including the genes for cyclin D1 and the c-Myc gene.9, 10 Accordingly, the gene for β-catenin is considered to be a type of oncogene. By contrast, apc and axin1, which participate in the degradation of β-catenin, appear to function, in this context, as tumor suppressors.
Mutations in the genes for β-catenin, apc, and axin1 have been identified in various neoplasms. Mutations in CTNNB1, the gene for β-catenin, are found in colon cancer, desmoid tumors, gastric cancer, hepatocellular carcinoma, medulloblastoma, melanoma, ovarian cancer, pancreatic cancer, and prostate cancer.2 Mutations in APC have frequently been identified in colon cancer.11 Mutations in AXIN1, the gene for axin1, have been found in hepatocellular carcinoma12 and medulloblastoma.13 Thus, there is considerable evidence for abnormalities in the Wnt signaling pathway in tumorigenesis.
According to the latest textbook of Armed Forces Institute of Pathology, adenoid cystic carcinoma is the fifth most common carcinoma of the minor and major salivary glands behind mucoepidermoid carcinoma; adenocarcinoma, not otherwise specified; acinic cell adenocarcinoma; and polymorphous low-grade adenocarcinoma.14 Adenoid cystic carcinoma is classified histologically into tubular, cribriform, and solid types. The general consensus is that adenoid cystic carcinoma with tubular or cribriform growth patterns (low grade) have a better prognosis than those with solid growth patterns (high grade).14
To our knowledge, while several earlier investigations have focused on genetic and biological alterations in adenoid cystic carcinoma, there is only one report that mentions the altered expression of genes in the Wnt signaling pathway.15 The goal of the present study was to examine possible abnormalities of CTNNB1, AXIN1, and APC genes in adenoid cystic carcinoma of the salivary gland.
Materials and methods
Materials
In all, 20 paraffin-embedded blocks of adenoid cystic carcinoma of the major and minor salivary gland were retrieved from among archival specimens at the Pathology Center of Oita University Hospital. There were six tubular-type adenoid cystic carcinomas, 10 cribriform-type adenoid cystic carcinomas, and four solid-type adenoid cystic carcinomas. When an adenoid cystic carcinoma appeared to be a combination of two or three subtypes, it was classified by reference to the dominant type. For each case, representative blocks, including the smallest amount of non-neoplastic tissue, were selected.
Extraction of DNA
Genomic DNA was extracted with DEXPAT™ (Takara, Tokyo, Japan) according to the manufacturer's instructions. In brief, 10 μm sections were cut from each block, transferred to sterilized test tubes and incubated with approximately 250 μl of DEXPAT™ at 100°C for 10 min. After centrifugation, the supernatant from each tube was collected and used as the source of the template for amplification by the polymerase chain reaction (PCR). Samples of DNA from peripheral leukocytes of healthy volunteers were used as controls.
PCR and Analysis of Single-Strand Conformational Polymorphism (SSCP)
Primers for amplification by PCR were designed to encompass exon 3 of CTNNB1, the mutation cluster region of APC,16 and the entire coding exon of AXIN1. The sequences of the various primers are shown in Tables 1, 2 and 3. Since each target of PCR could not be amplified by a single PCR because of the degraded state of the template DNA that had been extracted from paraffin-embedded tissues, we used several sets of primers to obtain amplified products of the entire length of each targeted DNA. We performed PCR in 25-μl reaction mixtures that contained 2.5 μl of a solution of template DNA solution, 1 × PCR buffer (Applied Biosystems, Foster City, CA, USA), 1.5 mmol/l MgCl2, 0.25 mmol/l dNTPs, 0.4 μmol/l primers, and 1 U Taq DNA polymerase (AmpliTaq Gold; Applied Biosystems). In total, 40 cycles of amplification were performed in a thermal cycler (9600; Applied Biosystems). The conditions for PCR were as follows: one cycle of denaturation at 94°C for 5 min, followed by 40 cycles of denaturation at 94°C for 30 s, annealing at 60°C for 30 s, and extension at 72°C for 30 s, with final extension at 72°C for 7 min.
For the analysis of single-strand conformational polymorphism (SSCP), products of PCR were denatured by heating in a solution of 50% formamide and 10 mM EDTA that contained bromophenol blue and xylene cyanol and separated by electrophoresis on a 12.5% polyacrylamide gel using a genophore™ system (Amersham Pharmacia, Upsalla, Sweden). Bands of DNA were visualized by silver staining with a genophore™ silver-staining kit plus (Amersham Pharmacia) according to the manufacturer's instructions.
DNA Sequencing
We determined the sequences of products of PCR and of plasmid vectors that included the products of PCR.
Purified products of PCR were subjected to direct sequencing with the forward and reverse primers specific for each product of PCR and BigDye™ Terminator Cycle Sequencing Ready Reaction Mix (Applied Biosystems). Purified products of PCR were ligated into the pCR vector and introduced into Escherichia coli with a TA cloning kit (Invitrogen, Carlsbad, CA, USA). Plasmid DNA isolated from colonies of Escherichia coli was sequenced with M13 reverse primer or the T7 universal primer and the BigDye™ Terminator Cycle Sequencing Ready Reaction Mix (Applied Biosystems). All sequencing was performed by an ABI PRISM 310 Genetic Analyzer (Applied Biosystems).
SSCP and these mutation analyses were repeated to eliminate PCR artifacts.
Immunohistochemical Staining
The antibody specific for β-catenin was purchased from Novocastra Laboratories Ltd (Newcastle, UK). Paraffin-embedded specimens of adenoid cystic carcinoma were cut at a thickness of 4 μm, deparaffinized in xylene, rehydrated, and autoclaved for 10 min in 10 mM citrate buffer, pH 6.0. Endogenous peroxidase activity was blocked using 0.3% hydrogen peroxide for 5 min. Sections were treated with the primary antibody at a 1:100 dilution for 60 min at room temperature. Products of immunoreactions were detected with EnVision+/HRP (Dako, Glostrup, Denmark).
Results
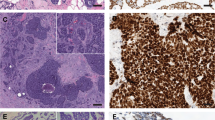
Representative results of SSCP analysis and the corresponding sequencing data are shown in Figure 1. The mutations that we detected and the results of immunohistochemical staining are summarized in Table 4. The base positions and codon numbers in the CTNNB1, AXIN1, and APC genes were determined from the NCBI database of Reference Sequences (RefSeq accession numbers NM 001904, NM 003502, and NM 000038, respectively). The first in-frame ATG triplet of each sequence is determined as codon 1, and position of the corresponding A residue is defined as position 1.

Examples of results of the analysis of single-strand conformational polymorphism and corresponding nucleotide sequencing (a), (b), and (c) show representative results for CTNNB1, APC, and AXIN1, respectively. Arrows of the side of electrophoregrams indicate polymorphism. T, tumor; N, normal control.
One case (case 16) included a mutation in CTNNB1, and this mutation caused residue Ile 35 to be replaced by Thr.
A mutation in APC was found in two cases (cases 14 and 18). One case included a point mutation that changed Arg 1399 to Cys. The other case revealed a deletion at base positions 4393 and 4394, which resulted in a change in the reading frame.
We detected seven missense mutations in AXIN1 in five cases (cases 6, 15, 16, 18, and 20), and two of these cases each included two mutations. The amino-acid substitutions were Gly 433 to Glu, Pro 661 to Leu, Glu 842 to Lys, Ser 828 to Gly, Phe 824 to Leu, Glu 842 to Gly, and Ser 828 to Tyr. The two cases with two mutations had substitutions of Glu 842 to Lys plus Ser 828 to Gly (case 16) and Glu 842 to Gly plus Ser 828 to Tyr (case 20). A previously reported single-nucleotide polymorphism was also detected in three cases (data not shown).
We examined possible correlations between mutations and histological type. Mutations were found in one of six cases of tubular type, in three of 10 cases of the cribriform type, and in three of four cases of the solid type.
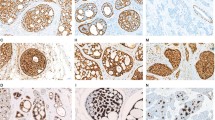
Figure 2 shows the results of immunostain for β-catenin in adenoid cystic carcinoma with specific growth pattern. Membranous expression of β-catenin was detected in the tumor cells in any type of growth pattern. Strong immunoreactions specific for β-catenin were detected in the nuclei of tumor cells in two cases (cases 16 and 18) that were associated with mutations identified in this study (Figure 3). One case was associated with mutations in both CTNNB1 and AXIN1 (case 16) and the other was associated with a mutation in APC (case 18). In all cases, we observed positive staining of cell membranes (Figure 2). Thus, the evaluation of very weak nuclear staining was difficult. We identified cases without evidence of strong immune reactions in the nuclei as immunonegative.

Adenoid cystic carcinoma with specific growth pattern and corresponding immunostain for β-catenin. The results of immunostain reveal membranous expression of β-catenin in tumor cells. (a, b) tubular pattern, (c, d) cribriform pattern, (e, f) solid pattern. (a), (c) and (e) Hematoxylin and eosin staining, (b), (d) and (f) corresponding immunostain for β-catenin.

Immunohistochemical detection of β-catenin in the nuclei. The nuclear expression of β-catenin is evident in several tumor cells.
Discussion
Adenoid cystic carcinoma is a generally slow-growing, but highly malignant neoplasm with a remarkable capacity for recurrence. Several investigations have found evidence of genetic and biological alterations in adenoid cystic carcinoma. Shintani et al17 demonstrated a high frequency of expression of c-erbB2 in adenoid cystic carcinoma. Papadaki et al18 reported that mutations in the tumor-suppressor gene TP53 are not important in early events in the development of adenoid cystic carcinoma but they involved at later stages of tumor progression and they are, thus, important in recurrence of adenoid cystic carcinoma. By contrast, Kiyoshima et al19 found infrequent mutations in TP53 in adenoid cystic carcinoma. It appears, therefore, mutation of TP53 does not necessarily play a critical role in the tumorigenesis of adenoid cystic carcinoma. Mutations in K-ras have been found in only a minority of cases of adenoid cystic carcinoma.20 The frequent expression of the c-kit protein has been noted in adenoid cystic carcinoma, but no mutations have been found in the c-kit gene in adenoid cystic carcinoma.21, 22
A relationship seems to exist between tumorigenesis and aberrations in the Wnt signaling pathway. Wnt signaling is crucial to embryonic development in a wide variety of organisms. By stimulating the transcriptional activities of DNA-binding proteins in the TCF/LEF-1 family, the Wnt signaling cascade is able to control cell behavior. The target genes of Wnt signaling include genes for proteins that drive the cell cycle and growth factors, for example, cyclin-D1, c-Myc, and vascular endothelial growth factor. Since β-catenin stimulates cell proliferation by stimulating the transcriptional activity of members of the TCF/LEF-1 family, inappropriately accumulated β-catenin acts similar to the product of an oncogene. By contrast, apc and axin1, which are required for the degradation of β-catenin, function as tumor suppressors.23 Mutations in CTNNB1, APC, and AXIN1 have been reported in various types of tumor. However, aberrations in these three genes in adenoid cystic carcinoma have not been explored to date. In this study, we investigated mutations in CTNNB1, AXIN1, and APC in adenoid cystic carcinoma and found that 35% of adenoid cystic carcinomas had mutations in at least one of these three genes. Since the position of the Ile35Thr substitution in β-catenin, detected in case 16, is within the site of phosphorylation by GSK-3β,23 such mutated β-catenin might escape phosphorylation and subsequent degradation. The mutation cluster region of APC is responsible for binding to β-catenin.2 In two cases with missense and a frame-shift mutation in APC (cases 14 and 18), degradation of β-catenin might be impaired. Axin1 binds to apc, GSK-3β, and β-catenin and functions as a regulator of the degradation of β-catenin.6 We detected the mutations in the central region and the carboxyl-terminal region of axin1 (cases 6, 15, 16, 19, and 20). Substitutions of amino acids in these regions might disturb the binding of axin1 to other components of the Wnt signaling pathway, resulting in the escape of β-catenin from degradation. Thus, in adenoid cystic carcinomas with aberrations in these components, β-catenin is likely to accumulate and to function inappropriately as an activator of transcription factors.
We detected the nuclear expression of β-catenin immunohistochemically in two cases (cases 16 and 18). There were mutations in both CTNNB1 and AXIN1 in case 16 and a mutation in APC in case 18. Cases with mutations in AXIN1 only did not show any evidence of strong nuclear expression of β-catenin. However, our negative immunohistochemical results do not eliminate the possibility of nuclear localization of β-catenin because weak nuclear staining was difficult to evaluate, as noted above. Frierson et al15 found no nuclear immuno-reactivity specific for β-catenin in adenoid cystic carcinomas, but the results of our immunohistochemical staining are not inconsistent with theirs. Frierson et al15 also reported strong expression of Frizzled-7, which encodes a receptor for Wnt proteins that might suppress the function of apc and, therefore, promote β-catenin signaling. In this study, we did not examine the expression of Frizzled-7 but it is likely that disturbances in several members of the Wnt signaling pathway play roles in carcinogenesis in some adenoid cystic carcinomas.
There appeared to be some correlation between histological subtype and the presence of mutations since mutations were observed in one of six tubular-type adenoid cystic carcinomas, three of 10 cribriform-type adenoid cystic carcinomas, and three of four solid-type adenoid cystic carcinomas. Mutations were detected more frequently in high-grade (solid) adenoid cystic carcinoma than in low-grade (tubular and cribriform) adenoid cystic carcinoma. However, the number of samples was small and there was no statistically significant difference between high- and low-grade adenoid cystic carcinoma (P=0.065; χ2-test).
In conclusion, it appears that mutations in the CTNNB1, APC, and AXIN1 genes, which are related to the Wnt signaling pathway, are quite common in adenoid cystic carcinoma. Our results suggest that abnormalities in the Wnt signaling pathway might play a role in the tumorigenesis and growth of adenoid cystic carcinoma.
References
Wodarz A, Nusse R . Mechanisms of Wnt signaling in development. Annu Rev Cell Dev Biol 1998;14:59–88.
Polakis P . Wnt signaling and cancer. Genes Dev 2000;14:1837–1851.
Krasnow RE, Wong LL, Adler PN . Dishevelled is a component of the frizzled signaling pathway in Drosophila. Development 1995;121:4095–4102.
Kühl M, Sheldahl LC, Park M, et al. The Wnt/Ca2+ pathway: a new vertebrate Wnt signaling pathway takes shape. Trends Genet 2000;16:279–283.
Yang-Snyder J, Miller JR, Brown JD, et al. A frizzled homolog functions in a vertebrate Wnt signaling pathway. Curr Biol 1996;6:1302–1306.
Ikeda S, Kishida S, Yamamoto H, et al. Axin, a negative regulator of the Wnt signaling pathway, forms a complex with GSK-3β and β-catenin and promotes GSK-3β-dependent phosphorylation of β-catenin. EMBO J 1998;17:1371–1384.
Sieber OM, Tomlinson IP, Lamlum H . The adenomatous polyposis coli (APC) tumour suppressor—genetics, function and disease. Mol Med Today 2000;6:462–469.
Liu C, Li Y, Semenov M, et al. Control of β-catenin phosphorylation/degradation by a dual-kinase mechanism. Cell 2002;108:837–847.
Nishida N, Fukuda Y, Komeda T, et al. Amplification and overexpression of the cyclin D1 gene in aggressive human hepatocellular carcinoma. Cancer Res 1994; 54:3107–3110.
He TC, Sparks AB, Rago C, et al. Identification of c-MYC as a target of the APC pathway. Science 1998;281:1509–1512.
Fodde R, Smits R, Clevers H . APC, signal transduction and genetic instability in colorectal cancer. Nat Rev Cancer 2001;1:55–67.
Taniguchi K, Roberts LR, Aderca IN, et al. Mutational spectrum of β-catenin, AXIN1, and AXIN2 in hepatocellular carcinomas and hepatoblastomas. Oncogene 2002;21:4863–4871.
Baeza N, Masuoka J, Kleihues P, et al. AXIN1 mutations but not deletions in cerebellar medulloblastomas. Oncogene 2003;22:632–636.
Ellis GL, Auclair PL . Adenoid cystic carcinoma. In: Rosai J (ed). Atlas of Tumor Pathology, Third Series, Fascicle 17, Tumors of the Salivary Glands. Armed Forces Institute of Pathology: Washington, DC, 1996, pp 203–216.
Frierson Jr HF, El-Naggar AK, Welsh JB, et al. Large scale molecular analysis identifies genes with altered expression in salivary adenoid cystic carcinoma. Am J Pathol 2002;161:1315–1323.
Nagase H, Nakamura Y . Mutations of the APC (adenomatous polyposis coli) gene. Hum Mutat 1993; 2:425–434.
Shintani S, Funayama T, Yoshihama Y, et al. Expression of c-erbB family gene products in adenoid cystic carcinoma of salivary glands: an immunohistochemical study. Anticancer Res 1995;15:2623–2626.
Papadaki H, Finkelstein SD, Kounelis S, et al. The role of p53 mutation and protein expression in primary and recurrent adenoid cystic carcinoma. Hum Pathol 1996;27:567–572.
Kiyoshima T, Shima K, Kobayashi I, et al. Expression of p53 tumor suppressor gene in adenoid cystic and mucoepidermoid carcinomas of the salivary glands. Oral Oncol 2001;37:315–322.
Yamamoto Y, Virmani AK, Wistuba II, et al. Loss of heterozygosity and microsatellite alterations in p53 and RB genes in adenoid cystic carcinoma of the salivary glands. Hum Pathol 1996;27:1204–1210.
Holst VA, Marshall CE, Moskaluk CA, et al. KIT protein expression and analysis of c-kit gene mutation in adenoid cystic carcinoma. Mod Pathol 1999;12:956–960.
Edwards PC, Bhuiya T, Kelsch RD . c-Kit expression in the salivary gland neoplasms adenoid cystic carcinoma, polymorphous low-grade adenocarcinoma, and monomorphic adenoma. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 2003;95:586–593.
Kikuchi A . Tumor formation by genetic mutations in the components of the Wnt signaling pathway. Cancer Sci 2003;94:225–229.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Daa, T., Kashima, K., Kaku, N. et al. Mutations in components of the Wnt signaling pathway in adenoid cystic carcinoma. Mod Pathol 17, 1475–1482 (2004). https://doi.org/10.1038/modpathol.3800209
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.3800209
Keywords
This article is cited by
-
Assessment of CTNNB1 gene mutations and β-catenin immunoexpression in salivary gland pleomorphic adenomas and adenoid cystic carcinomas
Virchows Archiv (2018)
-
Butyrate and bioactive proteolytic form of Wnt-5a regulate colonic epithelial proliferation and spatial development
Scientific Reports (2016)
-
Next-Generation Sequencing in Salivary Gland Basal Cell Adenocarcinoma and Basal Cell Adenoma
Head and Neck Pathology (2016)
-
Expression patterns of WNT/β-CATENIN signaling molecules during human tooth development
Journal of Molecular Histology (2014)
-
Basal cell adenocarcinoma of the salivary gland: a morphological and immunohistochemical comparison with basal cell adenoma with and without capsular invasion
Diagnostic Pathology (2013)
