
Abstract
We have observed an increasing number of autopsies on patients with chemotherapy-related complications. One complication is toxic leukoencephalopathy, which is due to a direct toxic effect of chemotherapeutic agents on the central nervous system white matter. Autopsies of four cases of toxic leukoencephalopathy were performed following standard protocols. The brain and spinal cord were examined routinely, and histological sections were taken for evaluation. We report here three patients with hematologic malignancies and one patient with metastatic carcinoma with chemotherapy-induced leukoencephalopathy. The first was a 56-year-old male treated with multiple chemotherapeutics for multiple myeloma. He presented with confusion and focal seizures with a rapid progression to coma and decerebrate posturing. The second was a 36-year-old male who developed mental status changes, ataxia and dysarthria following treatment for lymphoma. The third was a 16-year-old male who developed a profound peripheral and central neuropathy after chemotherapy treatment for T-cell acute lymphoblastic leukemia. The fourth was a 49-year-old female patient who was treated with multiple chemotherapeutics for Stage II breast carcinoma and subsequently developed visual acuity and field defects. The neuropathologic findings in these cases, although similar, varied in severity and distribution. The white matter was affected by severe myelin pallor, edema, and a prominent macrophage infiltrate in each of the cases. The location and extent of the central nervous system pathology correlated with the type and severity of clinical symptoms. These four cases, with their varied presenting symptoms, clinical courses, and degree of pathology, emphasize the importance of considering toxic leukoencephalopathy as an etiology of acute neurologic deterioration following high-dose chemotherapy.
Similar content being viewed by others

Main
Leukoencephalopathy describes the structural changes of cerebral white matter in which myelin suffers the most extensive damage. This white matter damage can be the result of genetic, metabolic, and vascular disorders, demyelinating diseases, infection, trauma, or hydrocephalus and toxin exposure. Toxic leukoencephalopathy, in particular, is white matter damage that results from exposure to a wide variety of toxic agents. These causative agents range from sources such as environmental toxins and drugs of abuse to the iatrogenic effects of therapeutic agents. Among the therapeutic agents that are known to result in toxic leukoencephalopathy are cranial radiation, immunosuppressive drugs, and chemotherapeutic agents.1 We present here a total of four cases of toxic leukoencephalopathy that occurred as a direct result of high-dose chemotherapy. Two of these cases are from autopsies that were performed in close time proximity at the Duke University Medical Center. They served as the impetus for this report. The two additional cases, which had previously been reported,2, 3 were included because of similar clinical and pathologic findings. It will be demonstrated that chemotherapy-induced toxic leukoencephalopathy can be manifested in a wide and diverse range of signs and symptoms. Consequently, it is imperative that both clinicians and pathologists maintain a high index of suspicion to facilitate an accurate and timely diagnosis.
Materials and methods
Autopsies were performed following Duke University Health System protocols. The brain and spinal cord were examined routinely and histologic sections were embedded in paraffin, cut at 8 μm and stained with hematoxylin and eosin/luxol fast blue. Selected sections were immunostained with antibodies to glial fibrillary acidic protein (GFAP), Ham56 (a macrophage marker), and neurofilament.
Results
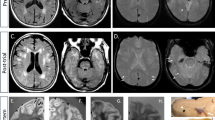
The first case was a 56-year-old white male who presented with bone pain, hypercalcemia, and proteinuria. A bone marrow biopsy confirmed a diagnosis of multiple myeloma. He was treated with eight cycles of vincristine, melphalan, cyclophosphamide, prednisone, BCNU, and adriamycin (VMCP/VBAP). Remission was not achieved, and consequently he underwent an autologous bone marrow transplant. During his transplant protocol, he received high doses of cyclophosphamide, etoposide, BCNU, and melphalan. He subsequently developed a chemotherapy-induced pre-B-cell acute lymphoblastic leukemia that was treated with gemcitabine and mitoxantrone. He also received a chimeric bone marrow transplant that involved treatment with campath, cytoxan, and fludarabine. At 3 weeks following this final chemotherapy regime, he began to experience confusion and a progressive deterioration of his mental status. He eventually developed wandering nystagmus, decerebrate posturing, and became unresponsive. An EEG performed during this time demonstrated findings consistent with diffuse encephalopathy. An MRI of his brain demonstrated a diffuse, nonenhancing, hyperintense T2-signal abnormality within the white matter of his frontal, parietal and temporal lobes, pons, corpus callosum, cerebral peduncles and corticospinal tracts (Figure 1). The patient suffered an aspiration event that led to increasing respiratory compromise and his eventual death. Post-mortem examination of his brain confirmed the previously described radiographic findings. Microscopic analysis revealed severe myelin pallor, edema, gliosis, and macrophage infiltrate of the deep cortical and cerebellar white matter, spinal cord, pons, and descending cortical spinal tract (Figures 2, 3 and 4).

Brain MRI (Case 1) demonstrating diffuse, nonenhancing, hyperintense T2-signal abnormality within the white matter of the frontal, parietal, and temporal lobes, pons, corpus callosum, cerebral peduncles, and corticospinal tracts.

LFA/H&E-stained sections of the cerebral cortical white matter show diffuse leukoencephalopathy in the form of myelin pallor, edema, vacuolization (Case 1).

GFAP immunostaining discloses numerous reactive astrocytes in cerebral cortical white matter (Case 1).

Ham-56 immunostaining discloses a prominent macrophage infiltrate in cerebral cortical white matter (Case 1).
The second case was a 36-year-old male who presented with bilateral cervical, epitrochlear, and inguinal adenopathy. This case has been previously reported.3 A lymph node biopsy confirmed a diagnosis of stage IVA Hodgkin lymphoma, mixed cellularity type. He was treated with vincristine, cisplatin, vinblastine, BCNU, procarbazine, bleomycin, and nitrogen mustard. He failed this initial chemotherapy regime and underwent an autologous bone marrow transplant that involved high-dose BCNU. Approximately 1 month following his final BCNU therapy, he began to develop progressive mental status changes, ataxia, and dysarthria. An EEG during this time demonstrated bilateral theta and delta activity with no normal alpha rhythms. A CT of his brain was normal. He developed multiple seizures and died 1 month subsequent to the onset of his neurologic symptoms.
Post-mortem examination of his brain demonstrated myelin pallor, edema, and necrosis predominantly in the temporal lobes and adjacent insular cortex (Figure 5). Neuropathologic findings in this case have been previously described.3 Briefly, there were focal areas of myelin pallor and vacuolization in the temporal lobes and adjacent insula. Larger areas of amorphous necrotic material were seen in the subcortical white matter. Throughout the brain stem, corpus callosum, cerebellum, and spinal cord, there were small discrete foci of myelin pallor with a bubbly appearance consistent with swollen axons. In the pons, these foci were seen in both the crossing and the descending fiber tracts. These foci also involved the pontine neurons in some areas (Figure 6).

LFA/H&E-stained sections demonstrating myelin pallor, edema, and necrosis predominantly in the temporal lobes and adjacent insular cortex (Case 2).

LFA/H&E-stained sections of the pons demonstrating focal areas of myelin pallor and vacuolization in the brain stem (Case 2).
The third case was a 16-year-old white male who was diagnosed with T-cell acute lymphoblastic leukemia. His treatment was initiated with the chemotherapeutic agents vincristine, doxyrubicin, methotrexate, 6-mercaptopurine, and prednisone. He failed this initial treatment and subsequently underwent a bone marrow transplant protocol that involved AraG, cytarabine, etoposide, and L-asparaginase therapy. Soon after his initial chemotherapy regime, he began to develop a peripheral neuropathy. He demonstrated a progressive weakness that was greater in the lower extremities than the upper and more profound distally than proximally. Eventually he was unable to ambulate.
An EMG study during this time demonstrated severe dysfunction of the motor and sensory nerves and extensive denervation in a predominantly distal pattern. These findings were noted to be most consistent with a severe, sensorimotor axonal polyneuropathy. An EEG demonstrated findings consistent with a severe diffuse neuronal dysfunction that was nonspecific in nature, and a brain MRI demonstrated a nonenhancing, hyperintense T2-signal abnormality within the pons, medulla, and dorsal columns of the cervical spines. The patient developed increasing respiratory distress that eventually led to his death. Post-mortem examination of the brain confirmed the MRI findings. Microscopic analysis demonstrated demyelinating lesions similar to the previously described cases. The lesions in this patient consisted of diffuse pallor in the posterior columns of the medulla. In the spinal cord, there was also severe pallor in the posterior columns. In addition, there was mild pallor and gliosis within the lateral spinal cord. Within the pons, there were punctuate areas of myelin pallor. Within these areas of pallor, there were swollen axons and gliosis (Figures 7 and 8).

LFA/H&E-stained sections of pons demonstrating myelin pallor, edema, vacuolization, gliosis, and a prominent macrophage infiltrate (Case 3).

LFA/H&E-stained sections of spinal cord show a diffuse and prominent myelin pallor and gliosis.
The fourth and final case was a 49-year-old female patient who was diagnosed with Stage II breast cancer. She was surgically resected and treated with four cycles of doxorubicin and cyclophosphamide. This initial treatment did not eradicate her cancer, and she later presented with parasternal metastases for which she was treated with two cycles of doxorubicin, methotrexate, and 5-fluorouracil that yielded only a partial response. Consequently, she underwent an autologous bone marrow transplant. The chemotherapeutic agents cyclophosphamide, cisplatin, and BCNU were used. At 2 months following this therapy, she developed a horizontal band visual defect in the left eye. Her visual acuity at that time was noted to be 20/40, and she had a normal fundoscopic examination. She had a progressive decline in visual acuity over the next 2 days to 4/200 in the left eye and 20/200 in the right eye. An MRI at this time demonstrated a subtle enhancement of the optic nerves bilaterally. Treatment with steroids succeeded in stabilizing her vision; however, she developed increasing respiratory compromise and refractory hypotension that led to her death. Similar to the previous cases, post-mortem examination of her brain demonstrated focal demyelination with edema, myelin pallor, macrophage infiltrate, and gliosis. This pathology was localized in the optic tracts and nerves, medulla, and posterior columns of the cervical spinal cord.2
Discussion
The four post-mortem examinations discussed here further illustrate many of the previously described clinical and neuropathologic features associated with toxic leukoencephalopathy.4 They well demonstrate that the clinical presentation of toxic leukoencephalopathy can be widely varied (Table 1). Despite the diverse presentation of these patients, however, they share common pathologic findings in the CNS white matter. They all have experienced a direct, chemotherapy-induced, toxic insult to the white matter. The common neuropathologic findings were of gliosis, macrophage infiltrate, and demyelinization of white matter.
The patients included in this study also confirm that mental status changes can be a prominent component of toxic leukoencephalopathy. Previously described mental status changes have been shown to range from mild conditions, such as chronic inattention and forgetfulness, to much more severe effects including coma and death.1, 5, 6, 7, 8 Cases 1 and 2 described above both experienced a decline in mental status as a result of their toxic leukoencephalopathy. The patient described in Case 3 experienced altered mental status. Unfortunately, his course was complicated by a drug-induced sedation. Although his altered mental status was temporally related to the chemotherapeutic insult, the use of sedatives during this time precludes ascertaining the specific etiology of his mental decline. Nevertheless, it is conceivable, in fact probable, that his chemotherapy and the resulting toxic leukoencephalopathy contributed, in part or in full, to his altered mental status.
Although mental status changes are more frequently encountered and reported as the major toxic effect of chemotherapy, the patients discussed here illustrate that toxic leukoencephalopathy can also present with very focal neurologic deficits. In fact, the effects may be so localized that chemotherapy toxicity is not even considered in the differential for an etiology. In Case 3, the patient presented with the focal findings of hemiparesis and sensory deficits. These clinical symptoms may be explained by the pathological demonstration of multiple demyelinating lesions within the pons, medulla, and lateral cortical spinal tracts. In addition, there was myelin pallor in the posterior columns that would correlate with the sensory deficits observed during life.
In Case 4, the patient presented with the focal finding of visual loss. At autopsy, demyelinating lesions in the optic chiasm were observed, which would explain this clinical symptom. In addition, small foci of demyelinating lesions were observed in the pons and medulla. However, these were much smaller than those observed in the previously described patient; and there were no clinically manifest symptoms. The lack of clinical correlation may be related to the size of the lesion.
Several chemotherapeutic agents, some of them used as treatment in the patients described here, have been previously described to result in toxic leukoencephalopathy. 1,3-bis(2-Chloroethyl)-1-nitrosourea (BCNU), a chemotherapeutic agent used in Cases 1, 2, and 4 presented here, is an alkylating agent. It has been available commercially since 1977 and has been used in bone marrow transplant protocols for several decades. During this time, several case reports of mental status changes and white matter degeneration have been described in association with its use. These symptoms have usually occurred within weeks of high-dose therapy.9 Melphalan, another alkylating agent that was used in Case 1, has had at least one report of two patients with toxic effects. These patients developed severe encephalopathy with seizures and mental status deterioration following treatment for multiple myeloma.10
Methotrexate (used in Cases 3 and 4) has a well-documented history of producing neurotoxic effects following administration. Within hours of a methotrexate dose, an aseptic meningitis has been shown to develop in some patients. A few days following administration, some patients have developed a toxic leukoencephalopathy with altered mental status and multifocal deficits. Within days to weeks, a transverse myelopathy with pain, sensory loss, paraplegia, and bladder dysfunction can develop. Months following treatment with methotrexate, some patients have demonstrated some cognitive impairment, dementia, spasticity, and seizure activity.11, 12, 13
Treatment with low doses of purine analogs such as fludarabine (used in Case 1), cytarabine (used in Case 3), and 5-fluorouracil (used in Case 4) has also previously been known to result in at least rare white matter changes. These effects have generally been mild and reversible. At higher doses such as in treatment for malignancies, however, more profound white matter changes have been documented by MRI with all three of these purine analog agents.1
The common chemotherapeutic agent, cisplatin (used in Cases 2 and 4), is effective due to its ability to bind DNA and inhibit repair. The most commonly documented neurologic effect of cisplatin is a peripheral neuropathy. Ototoxicity has also been reported. Rare cases of leukoencephalopathy with CNS white matter myelin destruction and necrosis have also been reported following high-dose therapy with cisplatin.1
Toxic leukoencephalopathy can also be seen in a variety of other settings that do not involve high-dose chemotherapy. Leukoencephalopathy has been seen in cases of cranial irradiation.14, 15 Indeed, cranial irradiation is the prototypical white matter toxin. It has been shown that the degree of neurotoxicity directly correlates with the total dosage of radiation. Of the patients receiving cranial irradiation, 28% demonstrate some degree of neurobehavioral dysfunction. This effect is potentiated in the setting of coexisting chemotherapy, particularly intrathecal therapy.12 Occupational exposures to organic solvents have been demonstrated to result in toxic leukoencephalopathy.16, 17, 18, 19, 20, 21, 22 Environmental toxins such as carbon monoxide, arsenic, and CCl4 have also been implicated.1 Drugs of abuse such as toluene, ethanol, cocaine, methamphetamine, and heroin have been associated with the phenomenon.23, 24, 25, 26, 27, 28, 29, 30, 31, 32 Even immunosuppressive agents such as amphotericin B and hexachlorophene have been shown to cause toxic leukoencephalopathy in patients.1
In addition to known toxic causes of leukoencephalopathy, there are several diseases that can present with a similar constellation of signs and symptoms. For example, genetic leukodystrophies and demyelinating diseases such as multiple sclerosis have been shown to have clinical manifestations similar to those of leukoencephalopathy. Additionally, some infectious agents can present with symptoms that could be readily misdiagnosed as toxic leukoencephalopathy. These include HIV infection in the form of an AIDS dementia complex, progressive multifocal leukoencephalopathy, subacute sclerosing panencephalitis, rubella, varicella-zoster, and cytomegalovirus. Various metabolic and vascular disorders have also been shown to have similar presentations. Even hydrocephalus and traumatic injuries have been implicated.1
The vast array of known causes and potential mimics of toxic leukoencephalopathy serve to remind us that it is important to maintain a broad differential when considering the diagnosis of toxic leukoencephalopathy. They also suggest that the number of yet undetermined causes is likely to be quite extensive. It is, therefore, prudent in all cases to consider other possible etiologic agents that must be ruled out prior to making a diagnosis of toxic leukoencephalopathy due to a chemotherapeutic agent.
Although many of the specific agents and mechanisms of toxic leukoencephalopathy have yet to be elucidated, the treatment and prognosis is fairly well understood. The prognosis depends largely on the extent of white matter injury and whether that injury is reversible or irreversible. Several case reports of reversible leukoencephalopathy with removal of the offending agent have been identified.1 Certainly, early recognition of the offending agent is crucial to facilitate timely termination while only minimal damage has occurred. Unfortunately, in the four cases reported here, early withdrawal was not possible because the onset of symptoms did not occur until after the completion of therapy. In light of the fact that this may often be the case, several therapeutic interventions to manage toxic leukoencephalopathy have been explored.1, 33, 34 Among these are corticosteroids, anticoagulation, ventriculoperitoneal shunting for leukotoxicity from cranial irradiation, and leucovorin for methotrexate toxicity. These treatments have demonstrated varying degrees of success.
The patients described in this report all experienced toxic leukoencephalopathy as a result of receiving high-dose chemotherapy. Although numerous antineoplastic agents have been implicated as the etiologic agents of toxic leukoencephalopathy, in none of the cases discussed here was toxic leukoencephalopathy considered as an etiology for the patient's symptoms. It was only after death that the pathology was demonstrated. These findings illustrate the fact that both clinicians and pathologists alike should maintain a high index of suspicion to correctly diagnose chemotherapy-associated toxic leukoencephalopathy in a timely manner.
References
Filley CM . Toxic leukoencephalopathy. Clin Neuropharmacol 1999;22:249–260.
Rubin P, Hulette C, Khawly JMI, et al. Ocular toxicity following high dose chemotherapy and autologous bone marrow transplantation: case report and pathological correlation. Bone Marrow Transplant 1996;18:253–256.
Burger PC, Kamenar E, Schold C, et al. Encephalomyelopathy following high-dose BCNU therapy. Cancer 1981;48:1318–1327.
Filley C, Kleinschmidt-DeMasters BK . Toxic leukoencephalopathy. N Engl J Med 2001;345:425–432.
Filley C, Kleinschmidt-DeMasters BK . Neurobehavioral aspects of cerebral white matter disorders. In: Fogel BS, Schiffer RB, Rao SM (eds). Neuropsychiatry. Williams & Wilkins: Baltimore, 1996, pp. 913–933.
Filley CM . The behavioral neurology of cerebral white matter. Neurology 1998;50:1535–1540.
Filley CM . Neurobehavioral anatomy. University Press of Colorado: Niwot, 1995.
Folstein MF, Folstein SE, McHugh PR . Mini-Mental State: a practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res 1975;12:189–198.
Brashir R, Hochberg FH, Linggood RM, et al. Pre-irradiation internal carotid artery BCNU in treatment of glioblastoma multiforme. J Neurosurg 1988;68:917–919.
Lebrun C, Chanalet S, Frenay M, et al. Leukoencephalopathy in multiple myeloma: 2 case reports. Ann Oncol 1999;10:1515–1517.
Mahoney DH, Shuster JJ, Nitschke R, et al. Acute neurotoxicity in children with B-precursor acute lymphoid leukemia: an association with intermediate-dose intravenous methotrexate and intrathecal triple therapy – a Pediatric Oncology Group study. J Clin Oncol 1998;16:1712–1722.
Asato R, Akiyama Y, Ito M, et al. Nuclear magnetic resonance abnormalities of the cerebral white matter in children with acute lymphoblastic leukemia and malignant lymphoma during and after central nervous system prophylactic treatment with intrathecal methotrexate. Cancer 1992;70:1997–2004.
Mulhern RK, Reddick WE, Palmer SL, et al. Neurocognitive deficits in medulloblastoma survivors and white matter loss. Ann Neurol 1999;46:834–841.
Sheline GE, Wara WM, Smith V . Therapeutic irradiation and brain injury. Int J Radiat Oncol Biol Phys 1980;6:1215–1228.
Crossen JR, Garwood D, Glatstein E, et al. Neurobehavioral sequelae of cranial irradiation in adults: a review of radiation-induced encephalopathy. J Clin Oncol 1994;12:627–642.
Rosenberg NL . Neurotoxicity of organic solvents. In: Rosenberg NL (ed). Occupational and Environmental Neurology. Butterworth-Heinemann: Boston, 1995, pp 71–113.
MMWR Organic solvents in the workplace. Morb Mortal Wkly Rep 1987;36:282–283.
Arlien-Soborg P, Bruhm P, Gyldensted C, Melgaard B . Chronic painters' syndrome: chronic toxic encephalopathy in house painters. Acta Neurol Scand 1979;60:149–156.
Baker EL . A review of recent research on health effects of human occupational exposure to organic solvents: a critical review. J Occup Med 1994;36:1079–1092.
Lees-Haley PR, Williams CW . Neurotoxicity of chronic low-dose exposure to organic solvents: a skeptical review. J Clin Psychol 1997;53:699–712.
Gamble JF . Low-level hydrocarbon solvent exposure and neurobehavioural effects. Occup Med (Lond) 2000;50:81–102.
Thuomas K-A, Moller C, Odkvist LM, et al. MR imaging in solvent-induced chronic toxic encephalopathy. Acta Radiol 1996;37:177–179.
Hormes JT, Filley CM, Rosenberg NL . Neurologic sequelae of chronic solvent vapor abuse. Neurology 1986;36:698–702.
Rosenberg NL, Kleinschmidt-DeMasters BK, Davis KA, et al. Toluene abuse causes diffuse central nervous system white matter changes. Ann Neurol 1988;23:611–614.
Kornfeld M, Moser AB, Moser HW, et al. Solvent vapor abuse leukoencephalopathy: comparison to adrenoleukodystrophy. J Neuropathol Exp Neurol 1994;53:389–398.
Filley CM, Heaton RK, Rosenberg NL . White matter dementia in chronic toluene abuse. Neurology 1990;40:532–534.
Bartzokis G, Goldstein IB, Hance DB, et al. The incidence of T2-weightedMR imaging Misc: signal abnormalities in the brain of cocaine-dependent patients is age-related and region-specific. AJNR Am J Neuroradiol 1999;20:1628–1635.
Bertram M, Egelhoff T, Schwarz S, et al. Toxic leukoencephalopathy following ‘ecstasy’ ingestion. J Neurol 1999;246:617–618.
Ricaurte GA, Forno LS, Wilson MA, et al. (+/−)3,4−Methylenedioxymethamphetamine selectively damages central serotonergic neurons in nonhuman primates. JAMA 1988;260:51–55.
Sprague JE, Everman SL, Nichols DE . An integrated hypothesis for the serotonergic axonal loss induced by 3,4-methylenedioxymethamphetamine. Neurotoxicology 1998;19:427–441.
Ginsberg MD, Hedley-Whyte ET, Richardson Jr EP . Hypoxic–ischemic leukoencephalopathy in man. Arch Neurol 1976;33:5–14.
Spengos K, Schwartz A, Hennerici M . Multifocal cerebral demyelination after magic mushroom abuse. J Neurol 2000;247:224–225.
Ghatak NR, Leshner RT, Price AC, et al. Remyelination in the human central nervous system. J Neuropathol Exp Neurol 1989;48:507–518.
Brustle O, Jones KN, Learish RD, et al. Embryonic stem cell-derived glial precursors: a source of myelinating transplants. Science 1999;285:754–756.
Acknowledgements
This work was supported by NIA AG05128 to Dr Hulette. We are grateful to Anne Latham for assistance with preparation of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Moore-Maxwell, C., Datto, M. & Hulette, C. Chemotherapy-induced toxic leukoencephalopathy causes a wide range of symptoms: a series of four autopsies. Mod Pathol 17, 241–247 (2004). https://doi.org/10.1038/modpathol.3800049
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.3800049
Keywords
This article is cited by
-
Allogeneic HSCT for Symptomatic Female X-linked Chronic Granulomatous Disease Carriers
Journal of Clinical Immunology (2023)
-
Erlotinib-induced leukoencephalopathy mimicking CO intoxication
Neurological Sciences (2022)
-
Investigation of chemotherapy-induced brain structural alterations in breast cancer patients with generalized q-sampling MRI and graph theoretical analysis
BMC Cancer (2018)
-
Chemotherapy and the pediatric brain
Molecular and Cellular Pediatrics (2018)
-
Recovery from chemotherapy-induced white matter changes in young breast cancer survivors?
Brain Imaging and Behavior (2018)
