
Abstract
We report two cases of intraneural synovial sarcoma. The first patient is a 46-year-old female who presented for several months with soft-tissue mass in the right infra-auricular region. The second patient is a 11-year-old girl who fell and then presented with pain in the area innervated by the right C7 spinal root and a nodule identified in the nerve root foramina. Both lesions were of small size and presented with features of synovial sarcoma. A biphasic variant was found in case 1 and a monophasic variant was present in case 2. Immunohistochemical studies were performed to confirm the diagnosis, excluding the main differential diagnoses, namely schwannoma and malignant peripheral nerve sheath tumor. Ultrastructural study was performed in case 2 allowing exclusion of other possible diagnoses. Molecular studies were performed on paraffin-embedded tissue in both cases and revealed the known characteristic t(X;18)(SYT-SSX) translocation.
Similar content being viewed by others


Main
Synovial sarcoma (SS) occurs primarily (>80%) in the periarticular regions of the extremities, usually in close association with tendon sheaths, bursae or joint capsules. Less than 20% of cases of SS occur away from the extremities, such as in the head and neck, the chest or the abdominal wall.1 Rare cases of SS have been identified in organs without any apparent relationship to synovial structures, such as the pleura,2, 3, 4 heart,5, 6, 7, 8 kidney,9, 10 prostate,11 intravascular spaces,12, 13 penis,14, 15 gastrointestinal tract,16, 17 parotid gland18 and vulva.19
To the best of our knowledge, there have only been five cases of intraneural synovial sarcoma (INSS) reported in the literature.20, 21, 22, 23, 24 Three of these cases were seen in teenagers (all 16 years old), while one occurred in a 23-year-old female patient and one occurred in a 43-year-old man. All patients presented with a subcutaneous soft-tissue mass involving major nerve trunks (the popliteal nerve,23 common digital nerve,20 median nerve22 and radial nerve24) often near body surfaces21 with a frequent history of a previous trauma. Four of the five tumors exhibited histologic and immunohistochemical features of a biphasic SS20, 21, 22, 23 and one was a monophasic SS.24 Molecular studies were not performed in these cases.
With ancillary techniques, a reciprocal chromosomal translocation t(X;18)(SYT-SSX) has been shown to be characteristic of SS.25 This translocation has been identified in over 90% of cases of SS using a reverse transcription-polymerase chain reaction (RT-PCR) assay.26, 27, 28 This molecular assay has been successfully applied to archival formalin-fixed, paraffin-embedded tumor tissue materials29 as well as to archival cytologic specimens.30
We describe two cases of primary INSS. One tumor was biphasic with epithelioid and spindled cells and the second one was monophasic with spindled cells only. Both were retained within the epineurium without extension into the perineural tissues. Both tumors showed the t(X;18)(SYT-SSX) by RT-PCR assay using formalin-fixed, paraffin-embedded tissue blocks, providing confirmation of the diagnosis.
Materials and methods
For light microscopic analysis, formalin-fixed and paraffin-embedded tumor tissue blocks were obtained. Sections, 4 μm thick, were cut for routine hematoxylin and eosin (H&E) stain and for immunohistochemistry. The commercially available primary anti-keratin 7 (1:2000, Dako, Carpinteria, CA, USA), keratin 19 (1:1600, Boehringer Mannheim, Indianapolis, IN, USA), epithelial membrane antigen (EMA) (1:1600, Dako), S-100 (1:2000, Dako) and vimentin (1:100, Ventana, Tucson, AZ, USA) and CD99 (1:160, Signet Laboratories, Inc., Dedham, MA, USA) antibodies were used in this study. Sections were deparaffinized and rehydrated in graded alcohol series. For heat-induced epitope retrieval (HIER), the sections were subjected to 100 mM EDTA buffer (pH 8.0) in a Steamer (Black & Decker, Shelton, CT, USA) at 100°C for 20 min. The sections were then brought to a Dako autostainer (Dako) following the vendor's protocol. Avidin–biotin complex (ABC) and peroxidase methods were used.
Ultrastructural analysis on case 2 was performed on a fragment of tumour tissue appropriately fixed in glutaraldehyde, embedded in Epon and examined under a Philips 300 electron microscope.
Molecular analysis was performed as previously described.30 Briefly, about 50 μg of tumor tissues were removed from each formalin-fixed and paraffin-embedded tissue blocks, deparaffinized in xylene and rehydrated in ethanol. After incubation in 250 μl of lysis buffer (20 mM Tris-HCl; 20 mM EDTA; 1% SDS; 350 μg proteinase K) overnight at 55°C, RNA was extracted with 750 μl of Trizol-LS (Life Technologies, Gaithesburg, MD, USA). RT-PCR was performed using a set of consensus primers that were designed to amplify both junctional regions of the SYT-SSX1 and SYT-SSX2 gene transcripts.30 Coamplification of β-actin mRNA was used as an internal positive control. The RT-PCR procedure was performed at least twice for each sample. The reaction products were subjected to electrophoresis in 2% agarose gel and visualized by ethidium bromide staining. The size of PCR products was 97 bp for the SYT-SSX fusion gene transcripts and 274 bp for the β-actin mRNA.
Clinical history
Case 1
A 46-year-old white female subject had a several month history of a mass (1.0 cm by imaging study) growing over the right infra-auricular region. The mass was not painful and originally started out like a grain of sand and gradually increased in size. The mass was excised as a local procedure in her physician's office and no margin was identified. The patient developed a hematoma immediately postexcision. This hematoma and surrounding tissue were widely resected in the following month. The patient received a complete course of radiation therapy to the right neck region after the resection. There was no residual sarcomatous tissue identified in the re-excised specimen. The patient has no clinical history of neurofibromatosis or other sequelae of neurofibromatosis type 1 (café-au-lait macules, optic glioma, iris hamartoma and sphenoid bony changes). After 5 years of follow-up, this patient is still free of recurrence.
Case 2
Following a fall while skiing, an 11-year-old white girl complained of pain in the territory innervated by the right C7 nerve root. She developed progressive weakness in the right triceps as well as increasing pain in the following days. On examination, she had complete atrophy/hypoplasia of the right scapular region as well as of the biceps and triceps. Right M4 triceps paresis and areflexia were observed. There was no pyramidal sign. An MRI revealed an expansile lesion (0.5 cm by imaging study) at the right C6–C7 level foramina, thought clinically to be a schwannoma. Complete surgical excision of the lesion was performed via a right C6–C7 laminectomy and foraminotomy 1 month after the onset of the symptoms. The postsurgical period was uneventful with complete disappearance of the pain. Proton therapy was then given. At 3 years after the first surgery and localized radiotherapy, a recurrence in the C7 nerve root and inferior trunk of the brachial plexus occurred and a second resection was performed for decompression and pain relief. The patient has no clinical history of neurofibromatosis or other sequelae of neurofibromatosis type 1 (café-au-lait macules, optic glioma, iris hamartoma and sphenoid bony changes). After 4 years of follow-up, this patient is still free of recurrence.
Results
Pathologic Findings
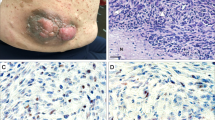
The material submitted to pathology was of small size, measuring 0.8 cm (case 1) and 0.4 cm (case 2) in greatest diameter in the resected specimens. They were both well-circumscribed, but unencapsulated and the cut surfaces were rubbery white-yellow. At low magnification, the tumor masses were retained within the epineurium. The tumors appeared as expansile masses infiltrating or pushing the nerve bundles apart (Figure 1). Case 1 consisted of a proliferation of tightly packed epithelioid and spindled cells with the latter predominating. The epithelioid cell component was comprised of large cuboidal to tall columnar cells with round or oval nuclei, thin chromatin, a distinctly outlined nuclear membrane and abundant pale-staining cytoplasm with well-demarcated cellular borders. These cells grew in solid cords, nests or arranged in pseudoglandular formations that contained homogenous eosinophilic secretions in their center. The other and predominant pattern was comprised of spindle cells in a storiform arrangement surrounding the epithelioid cells. These cells had a uniform appearance, with oval and hyperchromatic nuclei containing one or two small nucleoli and indistinct scanty cytoplasm. Microcalcifications were focally seen. Case 2 was composed of uniform small fusiform cells growing in sheets within the nerve fibers without any distinct pattern formed and accompanied by a few dilated small vessels. There were no myxoid or hyalinized areas, and no foci of necrosis or inflammation in either case. Rare mitotic figures of normal appearance were seen in both cases.

SS forms tumor masses within the nerve trunks (facial nerve in case 1 and radial nerve in case 2). Normal nerve bundles (N) are pushed to the periphery. Tumor masses and nerve bundles are circumscribed by the epineurium (arrowheads). There are microcalcifications (right lower field in case 1) and engorged vessels (middle field of cases 2).
Immunohistochemical Findings
The spindle cells were diffusely positive for vimentin and negative for S-100 protein in both cases. The epithelioid cells in case 1 were strongly positive for keratin 7, keratin 19 and EMA (Figure 2), while case 2 was entirely negative for these antigens. CD99 was focally positive in case 2, but negative in case 1. Neurofilament (not shown) and S-100 protein (Figure 2) demarcated the involved axonal segments that were pushed aside in both cases. EMA stain highlighted the perineurium in case 1.

Relationship of SS and nerve bundles in case 1. In the H&E stained section, nerve bundles (N) are pushed aside by the expansile SS. By immunohistochemical stains, epithelioid cells in SS are positive for cytokeratin 19 and EMA (inserts), and nerve bundles are positive for S-100 protein. EMA stain highlights perineurium (arrows).
Ultrastructural Study (case 2)
Appropriately fixed tumor tissue was analyzed for electron microscopy. It consisted of a uniform cellular proliferation, with small oval or round nuclei with slightly irregular contours, thin chromatin and small nucleoli. The cytoplasm was abundant and mainly elongated in which few organelles could be found (mitochondria, small rough endoplasmic reticulum and abundant intermediate filaments). These cells formed no junction between them. The intercellular spaces were scanty and a few collagen fibers were seen. No well-formed basal lamina or Luse's body was found around the tumor cells.
Molecular Findings
For detection of the t(X;18) (SYT-SSX) translocation using RT-PCR analysis, for each case, two PCR amplifications were performed in parallel. One contained only the primer set necessary for the amplification of SYT-SSX fusion gene transcripts, whereas in the other one a set of primers for the ubiquitously expressed β-actin mRNA was added. Both cases revealed evidence of the presence of a t(X;18) (SYT-SSX) translocation using RT-PCR (Figure 3).

Detection of the t(X;18) (SYT-SSX) translocation using RT-PCR analysis. For each case, two PCR amplifications were performed in parallel. One contained only the primer set necessary for the amplification of SYT-SSX fusion gene transcripts, whereas in the other one, a set of primers for the ubiquitously expressed β-actin mRNA was added. M, DNA size marker (100 bp DNA ladder); C−, negative control consisted of a t(X;18)-negative desmoid tumor; C+, positive control (biphasic SS); Case NT, neuronal tumor without t(X;18) translocation; Lanes 2,3,5,7,9,11,13,15, PCR for β-actin and synovial translocation t(X;18); Lanes 4,6,8,10,12,14, 16, PCR for synovial translocation t(X;18) only.
Discussion
The unusual location of an SS within a nerve root makes this diagnosis difficult without the use of specific ancillary techniques. There is no predominant histologic feature to confirm the diagnosis of an SS originating in the intraneural location unless a biphasic component (epithelioid cells and spindled cells) is present. Several spindle cell neoplasms may grow inside the nerve trunks, including perineurioma31, 32 and peripheral nerve sheath tumor.33, 34, 35 SS with a biphasic component may be easier to diagnose, but the monophasic presentation becomes a diagnosis by exclusion, and needs more sophisticated techniques, particularly molecular biology, to reach a proper diagnosis with certainty.
The spindle cell tumor most often encountered in nerve trunk is a peripheral nerve sheath tumor (PNST), including both the benign neurofibroma or schwannoma and the malignant peripheral nerve sheath tumor (malignant schwannoma). INSS and intraneural peripheral nerve sheath tumor may have overlapping histologic features, in that both tumors may be composed of spindled cells with epithelioid cells. Immunohistochemistry with cytokeratins and S-100 protein has been shown to be helpful in the differential diagnosis of biphasic SS vs PNST by demonstrating distinct cytokeratin 7 and 19 positive cells in biphasic SS and S-100 protein positivity in PNST.36, 37 PNST may occasionally contain glandular elements that may closely stimulate SS. However, these glandular elements histologically show mucinous features and frequently express neuroendocrine markers such as chromogranin and serotonin,38 whereas the epithelial elements in INSS are consistently nonreactive for these neuroendocrine markers.20
Like INSS, intraneural perineurioma is a rare clinical entity, which also tends to affect major nerve trunks.31, 32 Thus, intraneural perineurioma may be confused with intraneural monophasic SS. On light microscopic examination, the spindle cells in perineurioma are generally more thin and elongated than seen in SS, and form pseudo-onion bulb structures having a clear central zone. The immunohistochemical features of both tumors may be similar, with positivity for EMA and negativity for S-100 protein.31 By more sophisticated molecular technique study, the intraneural perineurioma has not been shown to have the t(X;18) translocation.
Molecular diagnosis has emerged as an important adjunct to the diagnosis of SS. The SYT-SSX fusion gene transcript has been detected in over 90% of cases of SS that were diagnosed based on conventional histology and immunohistochemistry.26, 27, 29, 30, 39 The SYT-SSX1 fusion transcript is more frequently seen in biphasic SS, whereas the SYT-SSX2 fusion transcript is more frequently seen in monophasic SS.28, 40 To make correct diagnosis of SS, one should combine the findings of molecular assays, immunohistochemistry and routine H&E morphology. In the current two cases, the SYT-SSX RT-PCR findings, histologic (biphasic and monophasic), immunohistochemical (positive for keratin and EMA and negative for S-100 in case 1) and electron microscopic (without evidence of PNST or perineurioma differentiation in case 2) features all supported the diagnosis of SS.
There are several recent reports demonstrating that many cases of renal embryonal sarcoma have the t(X;18) translocation.10, 41 In view of all these studies, it has been argued by Argani et al. that at least some cases of previously diagnosed renal embryonal sarcomas, adult blastemal/primitive stromal Wilms' tumors and adult cellular mesoblastic nephromas may in fact represent cases of renal monophasic SS;9 and a misleading diagnosis, because of the unusual location, may easily occur. In a similar vein, Billings et al. have also argued that gastrointestinal tract SS may be underdiagnosed because it may be confused with sarcomatoid carcinoma or gastrointestinal stromal tumor.16 Therefore, some ill-defined spindle cell sarcomas within various organ systems may be recognized as monophasic SS if more widespread molecular testing is utilized. To avoid confusion, the term synovial sarcoma itself in the future may need to be reconsidered.
Our cases of INSS have several similarities to the five previously reported cases (Table 1).20, 21, 22, 23, 24 Five of seven patients were female; six of them were teenagers or young adults (11, 16 and 23 years old). Compared to other soft-tissue sarcomas, all tumors were of relatively small size, ranging from 0.4 to 3.5 cm with an average size of 1.9 cm. Most of the tumors occurred in the major nerve trunks near exposed skin surface.
In summary, we are the first to report two cases of INSS with molecular evidence of specific t(X;18) (SYT-SSX) translocation. SS should be considered when dealing with an intraneural spindle cells lesion and molecular analysis for t(X;18) translocation should always be performed in such a situation.
References
Enzinger FM, Weiss SW . Synovial Sarcoma. In: Enzinger FM, Weiss SW (eds). Soft Tissue Tumors, 3rd edn. Mosby: St Louis, MO, 1995, pp 757–786.
Jawahar DA, Vuletin JC, Gorecki P, et al. Primary biphasic synovial sarcoma of the pleura. Res Med 1997;91:568–570.
Nicholson AG, Goldstraw P, Fisher C . Synovial sarcoma of the pleura and its differentiation from other primary pleural tumors: a clinicopathological and immunohistochemical review of three cases. Histopathology 1998;33:508–513.
Gaertner E, Zeren EH, Fleming MV, et al. Biphasic synovial sarcomas arising in the pleural cavity. A clinicopathologic study of five cases. Am J Surg Pathol 1996;20:36–45.
Casselman FP, Gillinov AM, Kasirajan V, et al. Primary synovial sarcoma of the left heart. Ann Thorac Surg 1999;68:2329–2331.
Kojima KY, Koslin DB, Primack SL, et al. Synovial sarcoma arising from the pericardium: radiographic and CT findings. Am J Radiol 1999;173:246–247.
Argani P, Askin FB, Colombani P, et al. Occult pulmonary synovial sarcoma confirmed by molecular technique. Pediatr Dev Pathol 2000;3:87–90.
Hisaoka M, Hashimoto H, Iwamasa T, et al. Primary synovial sarcoma of the lung: report of two cases confirmed by molecular detection of SYT-SSX fusion gene transcripts. Histopathology 1999;34:205–210.
Argani P, Faria PA, Epstein JI, et al. Primary renal synovial sarcoma. Molecular and morphologic delineation of an entity previously included among embryonal sarcoma of the kidney. Am J Surg Pathol 2000;24:1087–1096.
Kim DH, Sohn JH, Lee MC, et al. Primary synovial sarcoma of the kidney. Am J Surg Pathol 2000;24:1097–1104.
Fritsch M, Epstein JI, Perlman EJ, et al. Molecularly confirmed primary prostatic synovial sarcoma. Hum Pathol 2000;31:246–250.
Robertson NJ, Halawa MH, Smith MEF . Intravascular synovial sarcoma. J Clin Pathol 1998;51:172–173.
Miettinen M, Santavirta S, Slatis P . Intravascular synovial sarcoma. Hum Pathol 1987;18:1075–1077.
AL-Rikabi AC, Diab AR, Buckai A, et al. Primary synovial sarcoma of the penis. Scand J Urol Nephrol 1999;33:413–415.
Hiraga H, Nojima T, Isu K, et al. Histological and molecular evidence of synovial sarcoma of bone. J Bone Joint Surg 1999;81:558–563.
Billings SD, Meisner LF, Cummings OW, et al. Synovial sarcoma of the upper digestive tract: a report of two cases with demonstration of the X;18 translocation by fluorescence in situ hybridization. Mod Pathol 2000;13:68–76.
Habu S, Okamoto E, Toyosaka A, et al. Synovial sarcoma of the esophagus: report of a case. Surg Today 1998;28:401–404.
Grayson W, Nayler SJ, Jena GP . Synovial sarcoma of the parotid gland. A case report with clinicopathological analysis and review of the literature. S Afr J Surg 1998;36:32–34.
Nielsen GP, Shaw PA, Rosenberg AE, et al. Synovial sarcoma of the vulva: a report of two cases. Mod Pathol 1996;9:970–974.
O'Connell JX, Browne WL, Gropper PT, et al. Intraneural biphasic synovial sarcoma: an alternative "glandular" tumor of peripheral nerve. Mod Pathol 1996;9:738–741.
Cugola L, Pisa R . Synovial sarcoma: with radial nerve involvement. J Hand Surg 1985;10:243–244.
Chesser TJ, Geraghty JM, Clarke AM . Intraneural synovial sarcoma of the median nerve. J Hand Surg 1999;24:373–375.
Spielmann A, Janzen DL, O'Connell JX, et al. Intraneural synovial sarcoma. Skeletal Radiol 1997;26:677–681.
Rinehart GC, Mustoe TA, Weeks PM . Management of synovial sarcoma of the median nerve at the elbow. Plast Reconstr Surg 1989;83:528–532.
Clark J, Rocques PJ, Crew AJ, et al. Identification of novel genes, SYT and SSX, involved in the t(X;18)(p11.2;q11.2) translocation found in human synovial sarcoma. Nat Genet 1994;7:502–508.
Crew AJ, Clark J, Fisher C, et al. Fusion of SYT to two genes, SSX1 and SSX2, encoding proteins with homology to the Kruppel-associated box in human synovial sarcoma. EMBO J 1995;14:2333–2340.
Fligman I, Lonardo F, Jhanwar SC, et al. Molecular diagnosis of synovial sarcoma and characterization of a variant SYT-SSX2 fusion transcript. Am J Pathol 1995;147:1592–1599.
Kawai A, Woodruff JM, Healey JH, et al. SYT-SSX gene fusion as a terminant of morphology and prognosis in synovial sarcoma. N Engl J Med 1998;338:153–160.
Tsuji S, Hisaoka M, Morimitsu Y . Detection of SYT-SSX fusion transcripts in synovial sarcoma by reverse transcription polymerase chain reaction using archival paraffin-embedded tissue. Am J Pathol 1998;153:1807–1812.
Guillou L, Coindre J, Gallagher G, et al. Detection of the synovial sarcoma translocation t(X;18) (SYT;SSX) in paraffin-embedded tissues using reverse transcriptase-polymerase chain reaction: a reliable and powerful diagnostic tool for pathologists. A molecular analysis of 221 mesenchymal tumors fixed in different fixatives. Hum Pathol 2001;32:105–112.
Jazayeri MA, Robinson JH, Legolvan DP . Intraneural perineurioma involving the median nerve. Plast Reconstr Surg 2000;105:2089–2091.
Michalak S, Lefrancq T, Bertocchi C, et al. Intraneural perineurioma: an unusual nerve sheath tumor. Ann Pathol 1999;19:23–25.
Lallemand RC, Weller RO . Intraneural neurofibromas involving the posterior interosseous nerve. J Neurol Neurosurg Psychiatry 1973;6:991–996.
Camilleri IG, Milner RH . Intraneural lipofibroma of the median nerve. J Hand Surg [Br] 1998;23:120–122.
Mentzel T, Katenkamp D . Intraneural angiosarcoma and angiosarcoma arising in benign and malignant peripheral nerve sheath tumours: clinicopathological and immunohistochemical analysis of four cases. Histopathology 1999;35:114–120.
Smith TA, Machen SK, Fisher C, et al. Usefulness of cytokeratin subsets for distinguishing monophasic synovial sarcoma from malignant peripheral nerve sheath tumor. Am J Clin Pathol 1999;112:641–648.
Folpe AL, Schmidt RA, Chapman D, et al. Poorly differentiated synovial sarcoma. Immunohistochemical distinction from primitive neuroectodermal tumors and high-grade malignant peripheral nerve sheath tumors. Am J Surg Pathol 1998;22:673–682.
Christensen WN, Strong EW, Bains MS, et al. Neuroendocrine differentiation in the glandular peripheral nerve sheath tumor. Pathologic distinction from the biphasic synovial sarcoma with glands. Am J Surg Pathol 1988;12:417–426.
Inagaki H, Murase T, Otsuka T, et al. Detection of SYT-SSX fusion transcript in synovial sarcoma using archival cytologic specimens. Am J Clin Pathol 1999;111:528–533.
Antonescu CR, Kawai A, Leung DH, et al. Strong association of SYT-SSX fusion type and morphologic epithelial differentiation in synovial sarcoma. Diagn Mol Pathol 2000;9:1–8.
Faria PA, Argani P, Epstein JI, et al. Primary synovial sarcoma of the kidney: a molecular reappraisal of a subset of so-called embryonal renal sarcoma [abstract]. Mod Pathol 1999;12:94A.
Acknowledgements
We thank Dr Kevin Godfrey (Long Beach Community Hospital, Long Beach, CA, USA) for providing the block of case 1 and Dr. Louis Guillou (pathologist, Centre Hospitalier Universitaire Vaudois, Lausanne, Switzerland) for revising the histology of case 2.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Chu, P., Benhattar, J., Weiss, L. et al. Intraneural synovial sarcoma: two cases. Mod Pathol 17, 258–263 (2004). https://doi.org/10.1038/modpathol.3800048
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.3800048
Keywords
This article is cited by
-
Synovial sarcoma of the brachial plexus – a rare tumor in a rare area: a case report
Journal of Medical Case Reports (2018)
-
Monophasic synovial sarcoma presenting as a primary ileal mass: a case report and review of the literature
Journal of Medical Case Reports (2012)
-
Neuronal Differentiation of Synovial Sarcoma and Its Therapeutic Application
Clinical Orthopaedics and Related Research (2008)
-
Synovial sarcoma with radiological appearances of primitive neuroectodermal tumour/Ewing sarcoma: differentiation by molecular genetic studies
Skeletal Radiology (2006)
-
HMGA proteins in malignant peripheral nerve sheath tumor and synovial sarcoma: preferential expression of HMGA2 in malignant peripheral nerve sheath tumor
Modern Pathology (2005)
